Médico especialista do artigo

Novas publicações
hamartoma
Última revisão: 29.06.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
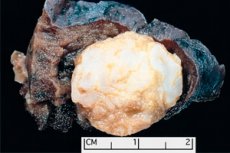
Formação tumoral localizada em qualquer região anatômica resultante do crescimento anormal de tecido benigno, em medicina é definida como hamartoma (do grego hamartia - erro, defeito). [ 1 ]
Epidemiologia
Estatisticamente, os hamartomas representam 1,2% das neoplasias benignas. A prevalência de hamartomas pulmonares é estimada em aproximadamente 0,25% da população em geral e representa até 8% de todas as neoplasias pulmonares. A maioria dos hamartomas pulmonares é diagnosticada incidentalmente em pacientes de 40 a 70 anos de idade, mas são muito raros na prática pediátrica.
Em geral, a maioria dos hamartomas é diagnosticada em homens, embora nos rins sejam mais comuns em mulheres e sejam identificados na meia-idade.
Cerca de 5% dos tumores benignos da mama são hamartomas e afetam mais comumente mulheres com mais de 35 anos.
80-90% das lesões hamartomatosas do cérebro e mais de 50% dos hamartomas do coração estão associados à esclerose tuberosa.
Causas hamartomas
Gamartomas pertencem a malformações congênitas e são formações de caráter benigno, formadas a partir de tecidos mesenquimais originários de folhetos germinativos. As causas de sua ocorrência estão associadas à divisão celular descontrolada de tecidos citologicamente normais (conjuntivo, músculo liso, gordura ou cartilagem), característicos de uma determinada localização anatômica, e ao seu crescimento focal excessivo durante a embriogênese de praticamente qualquer órgão ou estrutura anatômica.
A ocorrência de múltiplos hamartomas no mesmo paciente é frequentemente chamada de hamartomatose ou hamartoma pleiotrópico.
Esses tumores podem ocorrer esporadicamente ou na presença de certas doenças hereditárias autossômicas dominantes, bem como síndromes geneticamente determinadas.
Em muitos casos, os hamartomas formam-se quando uma doença genética rara de natureza multissistémica - esclerose tuberosa - se manifesta logo após o nascimento, ou na doença familiar de Recklinghausen - neurofibromatose tipo 1. [ 2 ]
Fatores de risco
Os principais fatores de risco para a formação de hamartomas incluem a presença das chamadas síndromes genéticas de polipose hamartomatosa na história dos pacientes, incluindo:
- Síndrome de hamartomas múltiplos - Síndrome de Cowden, na qual se formam múltiplos hamartomas de origem ecto, ento e mesodérmica, observam-se polipose gastrointestinal e manifestações mucocutâneas;
- Síndrome de Peutz-Jeghers-Turen (caracterizada pelo desenvolvimento de pólipos hamartomatosos benignos no trato gastrointestinal);
- Síndrome de Proteus;
- Síndrome de Weil - polipose juvenil do cólon;
- Síndrome de Bannayan-Riley-Ruvalcaba, que, assim como a síndrome de Cowden, produz múltiplos hamartomas (pólipos hamartomatosos) do intestino;
- Síndrome de Carney-Stratakis e complexo de Carney.
Além disso, hamartomas se formam em pacientes com síndrome de Watson hereditária e em casos de síndrome de Pallister-Hall esporádica ou congênita com hamartoma hipotalâmico e polidactilia.
Patogênese
O mecanismo de aumento da proliferação de tecidos germinativos com formação de malformações tumorais em vários órgãos é explicado por aberrações cromossômicas e mutações genéticas que podem ocorrer espontaneamente ou ser herdadas.
Na esclerose tuberosa, foram identificadas mutações nos genes TSC1 ou TSC2 – supressores tumorais que previnem e inibem a proliferação excessiva – crescimento e divisão celular muito rápidos ou descontrolados. E na neurofibromatose tipo 1 e na síndrome de Watson – mutações germinativas do gene supressor tumoral mitocondrial NF1.
Na síndrome do tumor hamartoma, que combina as síndromes de Cowden, Protea, Bannayan-Riley-Ruvalcaba e polipose juvenil, a patogênese está associada à mutação do gene PTEN, que codifica uma enzima envolvida na regulação da proliferação e é considerada um gene supressor de tumor.
Mutações no gene STK11, que codifica a estrutura e a função de uma das enzimas serina transmembrana, que reduz sua capacidade de restringir a divisão celular, levam à síndrome de Peutz-Jeghers-Turen, com o desenvolvimento de pólipos intestinais e lesões cutâneas pigmentadas. Uma mutação no gene GLI3, um fator de transcrição envolvido na formação do tecido intrauterino, foi identificada na síndrome de Pallister-Hall.
Assim, o crescimento celular descontrolado devido a mutações genéticas leva à formação de hamartomas.
Sintomas hamartomas
Dependendo da localização dos hamartomas, seus tipos são diferenciados, e cada um deles tem sua própria estrutura e sintomatologia.
Hamartoma do pulmão
O hamartoma pulmonar pode se formar em qualquer lobo e partes periféricas dos pulmões e consiste em tecidos normais presentes nos pulmões: adiposo, epitelial, fibroso e cartilaginoso. Em 80% dos casos, o componente condroide (células da cartilagem hialina) predomina, com inclusão de adipócitos – células do tecido adiposo e células epiteliais das vias aéreas. [ 3 ]
Os nomes anteriores: hamartoma condroide, mesenquimoma, hamartoma condromatoso ou hamartocondroma não são atualmente recomendados pela OMS.
O hamartoma cístico mesenquimal do pulmão, por outro lado, é menos comum e está associado à síndrome de Cowden na maioria dos pacientes.
A lesão hamartomatosa do pulmão pode não se manifestar, mas pode causar sintomas na forma de tosse crônica (frequentemente com hemoptise), chiado ao respirar e dificuldade para respirar. [ 4 ]
Um hamartoma do coração
Os tumores cardíacos primários benignos em adultos incluem o hamartoma de miócitos maduros e, em bebês e crianças com esclerose tuberosa, o rabdomioma, ou seja, o hamartoma miocárdico dos ventrículos ou do septo interventricular. [ 5 ]
O hamartoma de cardiomiócitos maduros se desenvolve na parede ventricular (e raramente nos átrios) e pode aparecer como lesões múltiplas, massas densas intimamente associadas ao miocárdio subjacente. O tumor pode causar sintomas de insuficiência cardíaca: dor no peito, palpitações e arritmias, sopros cardíacos, edema, dispneia e cianose.
Rabdomiomas cardíacos, a maioria dos quais é diagnosticada no primeiro ano de vida, são compostos de tecido muscular cardíaco formado por mioblastos embrionários e têm a aparência de massas focais sólidas sem cápsula.
Normalmente, esses hamartomas se apresentam de forma assintomática e regridem espontaneamente antes dos 4 anos de idade.
Lesões hamartomatosas também são consideradas por alguns especialistas como associadas ao mixoma cardíaco complexo de Carney. [ 6 ]
Gamartoma do trato gastrointestinal
Hamartoma gástrico é uma massa mesenquimal na forma de um pólipo hiperplásico epitelial do estômago, pólipo de Peutz-Jeghers e um raro hamartoma mioepitelial – com feixes de músculo liso hipertrofiados. Outros nomes para este hamartoma incluem hamartoma mioglandular, hamartoma adenomiomatoso e adenomioma gástrico. As manifestações clínicas típicas incluem dispepsia, dor epigástrica e sangramento gastrointestinal superior. [ 7 ], [ 8 ]
Mais informações no material - Polipose gástrica
Um hamartoma intestinal é um pólipo hamartomatoso ou hiperplásico do intestino grosso, diagnosticado como adenoma adenomatoso ou tubular. Quando o hamartoma está localizado na glândula de Brunner do duodeno, os sintomas se manifestam por dor na região epigástrica; náuseas, vômitos e flatulência (indicando obstrução intestinal); e, se de tamanho considerável, sangramento gastrointestinal. Em casos de hamartoma mioepitelial do íleo, os pacientes queixam-se de dor abdominal, apresentam diminuição do peso corporal e desenvolvem anemia crônica. [ 9 ], [ 10 ]
Leia também - Pólipos retais
Hamartoma retrorretal é um hamartoma cístico ou cisto multicâmara do espaço retrorretal (o tecido conjuntivo frouxo entre o reto e sua própria fáscia) que ocorre mais comumente em mulheres de meia-idade. Apresenta a aparência de um cisto que se projeta para fora da parede posterior do reto, revestida por epitélio e contendo fibras musculares lisas dispostas de forma caótica. Este hamartoma se manifesta com dor abdominal inferior e constipação recorrente. [ 11 ], [ 12 ]
Hamartomas do fígado e baço
Hamartoma biliar múltiplo do fígado é um hamartoma dos ductos biliares intra-hepáticos interdólicos associado a malformações do seu desenvolvimento durante o período embrionário. Este hamartoma (único ou múltiplo) consiste em aglomerados de ductos biliares e estroma fibrocolagenoso desordenadamente dilatados. [ 13 ]
Os hamartomas biliares são assintomáticos e geralmente são descobertos incidentalmente (durante exame radiológico ou laparotomia). [ 14 ]
Uma neoplasia primária de caráter benigno, rara e frequentemente detectada incidentalmente, é o hamartoma do baço, que consiste em elementos da polpa vermelha do baço – na forma de uma massa homogênea bem definida e de consistência firme. Essa malformação pode ser única ou múltipla; ao comprimir o parênquima esplênico, pode haver sensação de desconforto e dor na região subcostal esquerda. [ 15 ], [ 16 ]
Hamartomas renais
O hamartoma renal mais comum é diagnosticado como angiomiolipoma renal, pois esse tumor benigno consiste em tecido adiposo maduro com fibras musculares lisas e vasos sanguíneos incorporados. Forma-se na esclerose tuberosa em 40-80% dos casos. O aumento do tamanho do hamartoma (mais de 4-5 cm) leva à dor e ao aparecimento de sangue na urina. [ 17 ], [ 18 ]
Hamartoma da mama
As definições diagnósticas de hamartoma mamário aceitas pela OMS incluem termos como adenolipoma, condrolipoma e hamartoma mióide. Embora frequentemente chamado de fibroadenolipoma pelos mamologistas, a formação tumoral contém células de tecido fibroso, glandular e adiposo envoltas em uma fina cápsula de tecido conjuntivo com contornos distintos. Calcificações focais podem ser observadas à visualização. Neste caso, as manifestações clínicas estão ausentes. [ 19 ], [ 20 ]
Leia também - Tumores de mama
Hamartomas do cérebro
Um terço dos pacientes com esclerose tuberosa apresenta um hamartoma cerebral na forma de protuberâncias corticais intracranianas ou tubérculos em vários lobos – na fronteira entre a substância cinzenta e a branca – ou nódulos subependimários ao longo das paredes dos ventrículos cerebrais. Também pode ocorrer a formação de hamartoma astrocítico, um astrocitoma subependimário de células gigantes com ruptura cortical, neurônios dismórficos e grandes células gliais do parênquima cerebral (astrócitos). Os sintomas de hamartomas cerebrais incluem crises convulsivas e retardo mental em crianças. [ 21 ], [ 22 ]
Uma malformação rara que ocorre durante a embriogênese e está presente ao nascimento é o hamartoma hipotalâmico, que consiste em uma massa de neurônios heterotópicos e células gliais. À medida que o cérebro da criança cresce, o tumor aumenta de tamanho, mas não se espalha para outras regiões cerebrais. [ 23 ], [ 24 ]
Se tecidos hipertrofiados forem formados na parte anterior do hipotálamo (tubérculo cinéreo), onde a glândula pituitária se liga a ele, a malformação manifesta sintomas de desenvolvimento sexual prematuro central (antes dos 8-9 anos de idade): aparecimento de erupções cutâneas de acne, desenvolvimento precoce das glândulas mamárias e menarca precoce em meninas; pelos pubianos precoces e mutação da voz em meninos.
Quando os hamartomas se formam na parte posterior do hipotálamo, podem ocorrer anormalidades na atividade elétrica do cérebro, que na primeira infância se manifestam por convulsões e, em um estágio posterior (dos 4 aos 7 anos), por epilepsia com crises epilépticas focais com riso repentino ou com choro involuntário, crises atônicas e tônico-clônicas, bem como crises de agressão, problemas de memória e cognitivos.
Um hamartoma hipofisário é um adenoma hipofisário benigno que ocorre esporadicamente.
Adultos de meia-idade com síndrome de Cowden podem apresentar uma rara massa tumoral, um hamartoma cerebelar, diagnosticado como gangliocitoma cerebelar displásico ou doença de Lhermitte-Duclos. Os sintomas podem estar ausentes ou manifestar-se como cefaleia, tontura, coordenação motora prejudicada e paralisia de nervos cranianos individuais.
Hamartoma de linfonodo
Quando as células do músculo liso e do tecido adiposo, bem como os vasos sanguíneos e o estroma colagenoso dos linfonodos inguinais, retroperitoneais, submandibulares e cervicais crescem excessivamente, forma-se um hamartoma angiomiomatoso de um linfonodo ou hamartoma angiomiomatoso nodular - com substituição parcial ou completa de seu parênquima. [ 25 ], [ 26 ]
Um hamartoma da pele
Na presença de esclerose tuberosa ou neurofibromatose, são observados vários hamartomas da pele, mais frequentemente na forma de manchas hipopigmentadas; manchas de café e leite; angiofibroma (nas bochechas, queixo, sulcos nasolabiais); manchas shagreen de várias localizações (que são nevos do tecido conjuntivo); placas fibrosas na testa, couro cabeludo ou pescoço.
Uma manifestação dermatológica rara da esclerose tuberosa (especialmente em homens) é o hamartoma foliculocístico e de colágeno, caracterizado por abundante deposição de colágeno na derme, fibrose perifolicular concêntrica e cistos subcutâneos em forma de funil cheios de queratina observados no exame histopatológico. [ 27 ]
Aos hamartomas constituídos por melanócitos (células que produzem o pigmento melanina), a maioria dos especialistas também se refere a várias neoplasias melanocíticas, em particular os nevos melanocíticos congênitos, que representam uma anormalidade da embriogênese.
Em termos de etiologia, os hamartomas compostos de tecido vascular também são hemangiomas da pele.
Pacientes com síndrome de Peutz-Jeghers-Thuren apresentam hamartoma na forma de pigmentação irregular da pele e membranas mucosas - lentiginose periorificial
Casos de hamartoma ectodérmico-mesodérmico papular linear (Hamartoma moniliformis) apresentam uma erupção papular linear de cor de pele na cabeça, pescoço e parte superior do tórax.
E um hamartoma sebocítico é um hamartoma das glândulas sebáceas, leia mais na publicação - nevo sebáceo.
Hamartoma do olho
Lesões hamartomatosas pigmentadas da íris na neurofibromatose tipo 1 e na síndrome de Watson – na forma de aglomerados nodulares de melanócitos dendríticos – são definidas como hamartomas de íris ou nódulos de Lisch. São pápulas amarelo-acastanhadas, arredondadas, transparentes (geralmente não afetam a visão) e em formato de cúpula, que se projetam acima da superfície da íris.
E pacientes com angiofibroma juvenil da nasofaringe e polipose adenomatosa familiar freqüentemente desenvolvem hamartoma combinado da retina e epitélio pigmentar da retina - na forma de uma mancha preta na parte central (macular) da retina. [ 28 ]
Um hamartoma do nariz
O hamartoma nasal é definido por especialistas como hamartoma condromesenquimal nasal ou condroma nasal, devido à proliferação benigna do epitélio respiratório, glândulas submucosas e mesênquima condro-ósseo. Suas manifestações clínicas dependem do tamanho e da localização da lesão e incluem: congestão nasal, dificuldade na respiração nasal e na amamentação em bebês, secreção nasal aquosa e clara e sangramento nasal. Um hamartoma pode crescer com a criança e se espalhar para as órbitas oculares, resultando em deslocamento para frente ou para trás do globo ocular, estrabismo ou distúrbios oculomotores. [ 29 ]
Um hamartoma em uma criança
Todas as lesões hamartomatosas acima mencionadas de vários órgãos e estruturas anatômicas estão presentes em crianças com síndromes correspondentes.
Os recém-nascidos apresentam hamartoma mesenquimal da parede torácica ou hamartoma cartilaginoso da costela, que são massas sólidas imóveis resultantes do crescimento excessivo focal de elementos esqueléticos normais com elementos cartilaginosos, vasculares e mesenquimais. Este hamartoma pode causar insuficiência respiratória e o desenvolvimento da síndrome do desconforto respiratório. O hamartoma mesenquimal do fígado é o segundo tumor hepático benigno mais frequente em crianças. Esta formação semelhante a um tumor (mais frequentemente localizada no lobo direito do órgão) consiste em células do estroma mesenquimal, hepatócitos e células epiteliais do revestimento do ducto biliar. O quadro clínico inclui massa palpável na cavidade abdominal, anorexia e perda de peso e, em caso de tamanho significativo (até 10 cm ou mais), o tumor cobre os ductos biliares extra-hepáticos e a veia cava inferior, o que leva à icterícia e edema das extremidades inferiores.
Um hamartoma é um nefroma mesoblástico congênito (que ocorre em 1 em cada 200.000 bebês) que pode causar distensão abdominal no recém-nascido, com uma massa palpável de consistência densa no quadrante superior direito do abdômen. Os bebês também podem apresentar respiração rápida e superficial.
Anomalias congênitas raras incluem o hamartoma fibroso da infância, que ocorre em crianças nos primeiros dois anos de vida e se apresenta como uma massa nodular indolor nos tecidos subcutâneos da axila, pescoço, ombro e antebraço, costas e tórax, coxa, pé e genitália externa.
O hamartoma angiomatoso écrino em crianças pode estar presente ao nascimento ou manifestar-se na primeira infância. Este tumor benigno de natureza hamartomatosa geralmente apresenta a aparência de nódulos e/ou placas azuladas ou acastanhadas que resultam da proliferação de tecido de glândulas sudoríparas écrinas e capilares nas camadas média e profunda da derme. Este hamartoma pode causar hiperidrose localizada e aumento do crescimento de pelos.
Complicações e consequências
É consenso geral que os hamartomas raramente recorrem ou se transformam em tumores malignos. Frequentemente, apresentam poucos ou nenhum sintoma e, às vezes, até desaparecem com o tempo. Mas, em casos mais graves e dependendo do local de formação, essas malformações podem ter complicações e consequências graves.
Primeiro, um hamartoma pode crescer a tal ponto que começa a pressionar os tecidos e órgãos circundantes, interrompendo suas funções.
O hamartoma cardíaco em crianças pode levar a anormalidades persistentes do ritmo cardíaco, defeitos nas válvulas e comprometimento do fluxo sanguíneo intracardíaco, com subsequente insuficiência cardíaca congestiva.
As complicações dos pólipos hamartomatosos do trato gastrointestinal incluem sangramento gastrointestinal, obstrução e intussuscepção intestinal (com possível desfecho fatal). Um hamartoma renal grande pode provocar ruptura do rim.
Um hamartoma no cérebro pode causar síndrome de hidrocefalia obstrutiva.
Em hamartomas hipotalâmicos e hipofisários, a produção do hormônio somatotrópico (hormônio do crescimento) pode ser prejudicada, levando ao desenvolvimento de nanismo hipofisário (hipopituitarismo) em crianças. Hamartomas hipotalâmicos em crianças também podem levar à epilepsia resistente a medicamentos.
As complicações do hamartoma do epitélio pigmentar da retina são repletas de disfunção da retina e/ou do nervo óptico, edema macular, neovascularização da coroide e descolamento da retina.
Diagnósticos hamartomas
Uma parte importante do diagnóstico de hamartomas e síndromes relacionadas é a coleta de anamnese, incluindo histórico familiar.
Os exames laboratoriais incluem exames de sangue: clínica geral; eletrólitos séricos; perfil linfocitário; níveis de cálcio, potássio, fosfato e ureia; e testes de função hepática. Se possível, realiza-se uma biópsia por aspiração com agulha fina da massa, visto que o exame histológico é crucial no diagnóstico e na escolha das táticas de tratamento.
O diagnóstico instrumental fornece a visualização da formação semelhante a um tumor hamartomatoso e a identificação de sua localização exata, para a qual são utilizados raios X, angiografia, eletroencefalografia (EEG), ultrassom (sonografia), TC (tomografia computadorizada), PET (tomografia por emissão de pósitrons) e RNM (ressonância magnética).
Diagnóstico diferencial
Em qualquer massa anormal, o diagnóstico diferencial é muito importante. Assim, tuberculoma e hamartoma são diferenciados; hamartoma pulmonar e câncer de pulmão primário, carcinoide broncogênico, doença metastática. O hamartoma cerebral deve ser diferenciado de craniofaringioma e glioma hipotálamo-quiasmático. E o diagnóstico diferencial de hamartoma como nefroma mesoblástico congênito inclui tumor de Wilms (nefroblastoma maligno), sarcoma de células claras do rim e tumor renal ossificante em lactentes.
Quem contactar?
Tratamento hamartomas
Se o hamartoma for assintomático e for descoberto acidentalmente, não é necessário tratamento, mas é necessário monitorar seu "comportamento" e a condição do paciente. Em outros casos, a terapia visa reduzir a intensidade dos sintomas e prevenir complicações. Por exemplo, no caso de hamartoma hipotalâmico com sintomas de puberdade precoce, são prescritos certos medicamentos que inibem a liberação de certos hormônios. Medicamentos cardíacos são usados para tratar sintomas de insuficiência cardíaca em pacientes com hamartomas cardíacos.
A remoção cirúrgica dos hamartomas é indicada para confirmar o diagnóstico e em casos de sintomas intensos clinicamente incorrigíveis.
Por exemplo, hamartomas pulmonares podem ser ressecados por ressecção em cunha e, em casos graves, pela remoção de um lobo do pulmão (lobectomia). Um hamartoma mamário também pode ser excisado e, se for grande, pode ser necessária uma mastectomia parcial ou completa.
A termoablação por radiofrequência estereotáxica ou a ablação a laser podem ser utilizadas para remover pólipos hamartomatosos. A radiocirurgia com raios gama altamente focalizados – gamma knife para hamartomas hipotalâmicos ou hamartomas astrocíticos – também é utilizada.
Prevenção
O único método de prevenção do desenvolvimento de hamartomas pode ser considerado o rastreamento genético dos futuros pais da criança.
Previsão
O prognóstico geral desta anomalia congênita depende da localização e do tamanho da neoplasia, bem como das comorbidades e do estado geral de saúde do paciente.