Médico especialista do artigo

Novas publicações
Neuroblastoma em crianças: causas, diagnóstico, tratamento
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
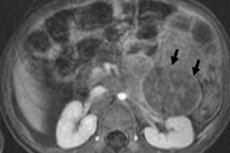
Em oncologia pediátrica, uma das neoplasias extracranianas mais comuns em crianças é o neuroblastoma, que é um tumor maligno embrionário dos neuroblastos da crista neural, ou seja, células nervosas embrionárias (imaturas) do sistema nervoso simpático.
Epidemiologia
De acordo com estatísticas do Grupo Internacional de Risco de Neuroblastoma (INRG), o neuroblastoma é responsável por cerca de 8% de todas as doenças oncológicas em crianças no mundo todo e é o terceiro em prevalência, depois da leucemia e dos tumores cerebrais.
De acordo com outros dados, o neuroblastoma representa cerca de 28% de todos os cânceres em bebês. Mais de um terço dos casos de neuroblastoma são diagnosticados em crianças menores de um ano; a idade média do diagnóstico é de 19 a 22 meses. Mais de 90% dos casos diagnosticados ocorrem em crianças de dois a cinco anos (com predominância de meninos); o pico de incidência é observado entre dois e três anos de idade, e os casos em crianças maiores de cinco anos representam menos de 10%.
Causas neuroblastomas
Ao estudar as causas do neuroblastoma, pesquisadores concluíram que esse tumor em crianças ocorre devido a mutações genéticas esporádicas durante a embriogênese ou no desenvolvimento pós-natal inicial. No entanto, a causa dessas alterações genéticas permanece desconhecida, uma vez que não foi identificada nenhuma influência de fatores ambientais teratogênicos.
Esses tumores podem ocorrer em qualquer lugar, incluindo mediastino, pescoço, abdômen, glândulas suprarrenais, rins, coluna e pelve.
Em casos raros, o neuroblastoma em bebês pode estar associado a uma mutação hereditária. Em particular, uma mutação no gene da proteína de membrana CD246 no cromossomo 2 – a enzima tirosina quinase ALK, que garante as comunicações intercelulares e desempenha um papel importante no funcionamento do sistema nervoso; e no gene da proteína PHOX2B (no cromossomo 4), que está envolvida na maturação das células nervosas.
O neuroblastoma também pode estar associado à neurofibromatose infantil tipo 1,à síndrome de Beckwith-Wiedemann e à hipoglicemia hiperinsulinêmica (pancreatite por nesidioblastose).
Fatores de risco
Hoje, a hereditariedade é reconhecida como fator de risco para o desenvolvimento de neuroblastoma em crianças – a presença desse tumor no histórico familiar, bem como anomalias congênitas associadas a mutações genéticas durante o desenvolvimento intrauterino. Isso é especialmente verdadeiro para casos de desenvolvimento de múltiplas neoplasias em diferentes órgãos.
Nenhum dos fatores exógenos que aumentam o risco desse tumor foi identificado pelos pesquisadores.
Patogênese
O mecanismo de desenvolvimento dos neuroblastomas é causado por distúrbios na diferenciação e maturação das células da crista neural – linhagens celulares bilaterais que se formam nas bordas do tubo neural a partir da camada germinativa ectodérmica do embrião humano. Essas células migram (se movem) e se diferenciam em diversos tipos de células: neurônios sensoriais e autônomos, células neuroendócrinas e células da medula adrenal, células da cartilagem e dos ossos craniofaciais, bem como células pigmentares.
No neuroblastoma, os neuroblastos migrados não amadurecem, mas continuam a crescer e se dividir, formando um tumor. E a patogênese de sua formação está associada às seguintes mutações genéticas:
- com duplicação de parte da sequência cromossômica ou duplicação de segmentos do gene LMO1 no cromossomo 11, que codifica a proteína RBTN1 nas células da crista neural do embrião;
- com uma alteração no número de cópias do gene NBPF10 no cromossomo 1q21.1, que codifica a proteína DUF1220, que controla a proliferação de células-tronco neurais humanas. Esses distúrbios levam à duplicação desse cromossomo ou à sua deleção – a ausência de parte do DNA;
- com alterações no gene supressor de tumor ATRX (no cromossomo Xq21.1);
- com a presença de cópias adicionais (amplificação) do gene do fator de transcrição N-Myc no cromossomo 2, que codifica um dos fatores de transcrição (proteína de ligação ao DNA) que regula a atividade de outros genes e controla a proliferação de células precursoras durante a formação de proteínas para a formação de tecidos e órgãos do feto. A amplificação desse gene o transforma em um oncogene, o que provoca a interrupção do ciclo celular, o aumento da proliferação celular e a formação de tumores.
Sintomas neuroblastomas
Os primeiros sinais do neuroblastoma são inespecíficos e podem incluir perda de apetite (e perda de peso), fadiga ao se alimentar, febre e dor nas articulações.
Os sintomas clínicos dependem da localização do tumor primário e da presença de metástases (que ocorrem em 60-73% dos casos).
Muito frequentemente, o neuroblastoma primário localiza-se na medula adrenal, que tem origem semelhante às células nervosas. Em crianças menores de um ano, o neuroblastoma adrenal é diagnosticado em 35-40% dos casos. Seus sintomas incluem dor abdominal, febre, perda de peso, dor óssea, anemia ou síndrome de Pepper concomitante: lesão hepática difusa com hepatomegalia grave e síndrome do desconforto respiratório.
O neuroblastoma retroperitoneal ou neuroblastoma retroperitoneal em crianças, à medida que cresce, começa a pressionar a bexiga ou os intestinos, o que pode causar problemas para urinar ou defecar, inchaço nas pernas (em meninos, o escroto incha).
O neuroblastoma do mediastino em crianças (neuroblastoma mediastinal) frequentemente pressiona a veia cava superior, o que pode causar inchaço na face, pescoço, braços e parte superior do tórax (com a pele ficando vermelho-azulada, com nódulos subcutâneos). Tosse e chiado no peito, problemas respiratórios (falta de ar) ou de deglutição (disfagia) aparecem; linfonodos aumentados são observados no pescoço, acima da clavícula e nas axilas.
A disseminação de células tumorais para a medula óssea leva à anemia, trombocitopenia e leucopenia, com tendência a sangramento.
E com metástases na região periorbitária, olheiras ou hematomas aparecem ao redor dos olhos. Esse tumor também pode causar dores de cabeça e tonturas, exoftalmia (protrusão dos globos oculares) e, devido à compressão das terminações nervosas, pálpebras caídas (ptose) e diminuição do tamanho das pupilas (miose).
O neuroblastoma abdominal ou neuroblastoma da cavidade abdominal em crianças leva à formação de selos palpáveis no abdômen, distensão abdominal, perda de apetite, constipação e aumento da pressão arterial. Um tumor que pressiona a medula espinhal ou a raiz nervosa pode causar dormência e fraqueza nos membros, além de incapacidade de ficar em pé, engatinhar ou andar. Se os ossos forem afetados, pode ocorrer dor óssea.
No caso de tumor em estágio 3-4 na cavidade abdominal com dano aos linfonodos, as células tumorais podem entrar no parênquima renal e, então, desenvolver-se um neuroblastoma extenso do rim em crianças, levando à interrupção de suas funções.
Estágios
- O neuroblastoma em estágio 1 é um tumor primário localizado e isolado em uma área do corpo; os gânglios linfáticos de ambos os lados não são afetados.
- Neuroblastoma estágio 2. No estágio 2A, o tumor primário está confinado a uma área, mas é grande; linfonodos bilaterais não estão envolvidos. No estágio 2B, os linfonodos do lado do corpo onde o tumor está localizado apresentam resultados positivos para metástases.
- Neuroblastoma estágio 3: o tumor primário cruza a medula espinhal ou a linha média do corpo, metástases unilaterais ou bilaterais são encontradas nos gânglios linfáticos.
- Neuroblastoma estágio 4: o tumor se espalhou para linfonodos distantes, medula óssea, ossos, fígado ou outros órgãos. Já o estágio 4S é determinado em crianças menores de um ano com tumor primário localizado, com disseminação para a pele, fígado ou medula óssea.
Sistema Internacional de Estadiamento de Risco de Neuroblastoma (INRGSS)
O INRGSS usa fatores de risco definidos por imagem (IDRFs), que são fatores observados em exames de imagem que podem significar que um tumor será mais difícil de remover.
O INRGSS divide os neuroblastomas em 4 estágios:
- L1: O tumor não se espalhou de onde se originou e não atingiu estruturas vitais. Está limitado a uma parte do corpo, como pescoço, tórax ou abdômen.
- L2: O tumor não se espalhou (metastatizou) para muito longe de onde começou (por exemplo, pode ter crescido do lado esquerdo do abdômen para o lado esquerdo do tórax), mas tem pelo menos um IDRF.
- M: O tumor sofreu metástase para uma parte distante do corpo (exceto para tumores no estágio de EM).
- EM: Doença metastática em crianças menores de 18 meses de idade, na qual o câncer se espalhou apenas para a pele, fígado e/ou medula óssea.
Complicações e consequências
O neuroblastoma é caracterizado por complicações e consequências como:
- disseminação (metástase) para os gânglios linfáticos, medula óssea, fígado, pele e ossos;
- compressão da medula espinhal (que pode causar dor e levar à paralisia);
- desenvolvimento da síndrome paraneoplásica (devido à ação de certas substâncias químicas secretadas pelo tumor, bem como do antígeno disialogangliosídeo GD2 expresso por suas células), que se manifesta por movimentos oculares involuntários rápidos, coordenação prejudicada, cãibras musculares e diarreia;
- recidivas após a conclusão da terapia primária (como mostra a prática clínica, neuroblastomas de alto risco apresentam recidiva em 50% dos casos).
Diagnósticos neuroblastomas
O diagnóstico de suspeita de neuroblastoma em uma criança requer exame, exames laboratoriais e de imagem.
São realizados exames de sangue e urina para detectar catecolaminas (norepinefrina e dopamina) e ácidos homovanílico ou vanililmandélico (formados durante o metabolismo desses hormônios); um exame de sangue para enolase neuroespecífica, um ensaio imunoenzimático (ELISA) do soro sanguíneo e uma análise da medula óssea (cuja amostra é coletada por punção aspirativa). Um teste de DNA é realizado para determinar mutações e uma biópsia é realizada para exame citomorfológico do tecido tumoral.
Após a coleta das amostras para biópsia, elas são enviadas para um laboratório, onde são analisadas ao microscópio por um patologista (médico com treinamento específico na identificação de células cancerígenas). Exames laboratoriais especiais também são frequentemente realizados nas amostras para verificar se o tumor é neuroblastoma.
Se for neuroblastoma, exames laboratoriais também podem ajudar a determinar a rapidez com que o tumor pode crescer ou se espalhar, bem como quais tratamentos podem funcionar melhor.
O diagnóstico instrumental visualiza a neoplasia por meio de ultrassonografia, raio-X, ressonância magnética ou tomografia computadorizada, PET com introdução de 18F-fluorodeoxiglicose ou varredura MIBG - cintilografia com metaiodobenzilguanidina. [ 1 ]
Diagnóstico diferencial
O diagnóstico diferencial inclui ganglioneuroma benigno, ganglioneuroblastoma, rabdomiossarcoma, nefroblastoma.
Quem contactar?
Tratamento neuroblastomas
No neuroblastoma, o tratamento depende do grupo de risco do paciente (estágio do processo tumoral), da localização do tumor, das características genômicas das células tumorais e da idade da criança. Pode incluir monitoramento, cirurgia, quimioterapia, radioterapia, imunoterapia e transplante de células-tronco hematopoiéticas.
A quimioterapia neoadjuvante ou adjuvante (pré ou pós-operatória) para neuroblastoma em crianças, assim como qualquer quimioterapia para câncer, é administrada em ciclos: o medicamento é administrado por vários dias consecutivos, seguidos de uma pausa para que o corpo se recupere. Os ciclos geralmente são repetidos a cada três ou quatro semanas.
Os seguintes medicamentos (e suas combinações) são usados: Ciclofosfamida, Cisplatina ou Carboplatina, Doxorrubicina (Adriamicina), Vincristina, Etoposídeo.
Os efeitos colaterais comuns dos medicamentos quimioterápicos incluem queda de cabelo, perda de apetite, fadiga, náuseas e vômitos, aftas, diarreia ou constipação. A quimioterapia pode danificar a medula óssea e causar diminuição da contagem de células sanguíneas.
A imunoterapia direcionada (direcionada ao antígeno tumoral GD2) utiliza medicamentos do grupo dos anticorpos monoclonais (anti-GD2 MAb), Dinutuximabe (Unituxin) e Naxitamabe. São administrados por via intravenosa por infusão prolongada, em combinação com fator estimulador de colônias de granulócitos-macrófagos (citocina GM-CSF) e interleucina-2.
Os efeitos colaterais desses medicamentos incluem dor (geralmente muito intensa), diminuição da pressão arterial, aumento da frequência cardíaca, falta de ar (com possível inchaço das vias aéreas), aumento da temperatura, náusea, vômito e diarreia, alterações na composição celular e mineral do sangue.
Para reduzir o risco de recorrência do câncer após quimioterapia de alta dose e transplante de células-tronco, crianças com neuroblastoma de alto risco são tratadas com retinoides sistêmicos, ácido 13-cis-retinóico (isotretinoína). [ 2 ]
Tratamento cirúrgico do neuroblastoma – remoção do tumor, por exemplo, adrenalectomia aberta ou ressecção laparoscópica do neuroblastoma adrenal; linfectomia (remoção dos gânglios linfáticos afetados), etc. [ 3 ]
Para neuroblastoma de alto risco, a radioterapia pode ser usada.[ 4 ]
Prevenção
Considerando as causas do neuroblastoma em crianças, a única medida preventiva pode ser o aconselhamento genético durante o planejamento da gravidez. No entanto, é importante lembrar que esse tumor está associado a mutações hereditárias em apenas 1% a 2% dos casos.
Previsão
O neuroblastoma infantil tem a capacidade de regredir espontaneamente.
Marcadores prognósticos
- Tumores de alto risco, assim como neuroblastoma em crianças de todas as faixas etárias e todos os estágios (exceto estágio 4S) – com aumento da expressão do gene N-MYC e amplificação do oncogene N-Myc – têm um prognóstico desfavorável que afeta a expectativa de vida.
- Ter células tumorais sem certas partes dos cromossomos 1 ou 11 (conhecidas como deleções 1p ou 11q) tem um prognóstico pior. Ter uma parte extra do cromossomo 17 (ganho de 17q) também está associado a um prognóstico pior.
- Células de neuroblastoma com grande quantidade de DNA têm melhor prognóstico, especialmente em crianças menores de 2 anos.
- Neuroblastomas que têm mais receptores de neurotrofina, especialmente o receptor do fator de crescimento nervoso TrkA, têm um prognóstico melhor.
Sobrevivência por grupo de risco do Childhood Oncology Group (COG)
- Grupo de baixo risco: Crianças no grupo de baixo risco têm uma taxa de sobrevivência de 5 anos superior a 95%.
- Grupo de risco intermediário: crianças no grupo de risco intermediário têm uma taxa de sobrevivência de 5 anos de 90% a 95%.
- Grupo de alto risco: crianças no grupo de alto risco têm uma taxa de sobrevivência de 5 anos de cerca de 50%.
Cerca de 15% das mortes por câncer infantil são causadas por neuroblastoma. As chances de sobrevivência a longo prazo para essa neoplasia maligna de alto risco não passam de 40%. A taxa geral de sobrevida em cinco anos é de 67% a 74%, sendo de 43% na faixa etária de um a quatro anos e superior a 80% para neuroblastoma diagnosticado no primeiro ano de vida.
Использованная литература