Médico especialista do artigo

Novas publicações
Doença de Charcot-Marie-Tooth.
Última revisão: 12.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
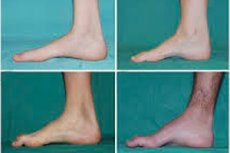
Atrofia muscular peroneal, síndrome ou doença de Charcot-Marie-Tooth, é um grupo de doenças hereditárias crônicas com danos aos nervos periféricos.
De acordo com a CID-10, na seção sobre doenças do sistema nervoso, o código para esta doença é G60.0 (neuropatia hereditária motora e sensorial). Também está incluída na lista de doenças raras.
Epidemiologia
De acordo com estatísticas clínicas, a prevalência de todos os tipos de doença de Charcot-Marie-Tooth por 100 mil habitantes é de 19 casos (de acordo com outras fontes, um caso por 2,5 a 10 mil habitantes).
A CMT tipo 1 representa cerca de dois terços dos casos (um caso para cada 5.000 a 7.000 habitantes), e quase 70% deles estão associados à duplicação do gene PMP22. Mais de 1,2 milhão de pessoas em todo o mundo sofrem desse tipo de doença.
A incidência de CMT tipo 4 é estimada em 1-5 casos por 10.000 crianças. [ 1 ]
Causas Doença de Charcot-Marie-Tooth
De acordo com a classificação das síndromes polineuropáticas, a atrofia muscular peroneal (fibular), a amiotrofia neural de Charcot-Marie-Tooth ou doença de Charcot-Marie-Tooth (abreviada como CMT) refere-se a polineuropatias motoras-sensoriais determinadas geneticamente. [ 2 ]
Ou seja, as causas de sua ocorrência são mutações genéticas. E dependendo da natureza das anormalidades genéticas, distinguem-se os principais tipos ou tipos dessa síndrome: desmielinizante e axonal. O primeiro grupo inclui a doença de Charcot-Marie-Tooth tipo 1 (CMT1), que ocorre devido à duplicação do gene PMP22 no cromossomo 17, que codifica a proteína 22 da mielina periférica transmembrana. Como resultado, ocorre a desmielinização segmentar da bainha do axônio (processos das células nervosas) e uma diminuição na velocidade de condução do sinal nervoso. Além disso, as mutações podem estar presentes em alguns outros genes.
A forma axonal é a doença de Charcot-Marie-Tooth tipo 2 (CMT2), que afeta os próprios axônios e está associada a alterações patológicas no gene MFN2 no locus 1p36.22, que codifica a proteína de membrana mitofusina-2, necessária para a fusão mitocondrial e a formação de redes mitocondriais funcionais dentro das células nervosas periféricas. Existem mais de uma dúzia de subtipos de CMT2 (com mutações em genes específicos).
Vale ressaltar que, atualmente, foram identificados mais de cem genes cujos danos, transmitidos por herança, causam vários subtipos da doença de Charcot-Marie-Tooth. Por exemplo, mutações no gene RAB7 levam à CMT tipo 2B; a alteração no gene SH3TC2 (que codifica uma das proteínas de membrana das células de Schwann) causa a CMT tipo 4C, que se manifesta na infância e é caracterizada pela desmielinização dos neurônios motores e sensoriais (há uma dúzia e meia de formas do tipo 4 desta doença).
Uma rara CMT tipo 3 (chamada síndrome de Dejerine-Sottas) começa a se desenvolver na primeira infância e é causada por mutações nos genes PMP22, MPZ, EGR2 e outros.
Quando o CMT tipo 5 ocorre entre 5 e 12 anos de idade, não apenas é observada neuropatia motora (na forma de paraparesia espástica das extremidades inferiores), mas também danos aos nervos óptico e auditivo.
Fraqueza muscular e atrofia óptica (com perda de visão), bem como problemas de equilíbrio, são típicos da CMT tipo 6. E na doença de Charcot-Marie-Tooth tipo 7, não há apenas neuropatia sensorial-motora, mas também uma doença da retina na forma de retinite pigmentosa.
Mais comum entre os homens, a CMT ligada ao cromossomo X ou doença de Charcot-Marie-Tooth com tetraparesia dos membros (fraqueza de movimento dos braços e pernas) é um tipo desmielinizante e acredita-se que resulte de uma mutação no gene GJB1 no braço longo do cromossomo X, que codifica a conexina 32, uma proteína transmembrana das células de Schwann e oligodendrócitos que regula a transmissão de sinais nervosos. [ 3 ]
Fatores de risco
O principal fator de risco para CMT é o histórico familiar da doença, ou seja, em parentes próximos.
Segundo geneticistas, se ambos os pais forem portadores do gene autossômico recessivo da doença de Charcot-Marie-Tooth, o risco de ter um filho que desenvolverá a doença é de 25%. E o risco de a criança ser portadora desse gene (mas não apresentar sintomas) é estimado em 50%.
No caso de herança ligada ao cromossomo X (quando o gene mutado está no cromossomo X da mulher), há 50% de risco de a mãe transmitir o gene ao filho, que desenvolverá CMT. A doença pode não ocorrer quando nasce uma menina, mas os filhos (netos) da filha podem herdar o gene defeituoso, e a doença se desenvolverá.
Patogênese
Em qualquer tipo de doença de Charcot-Marie-Tooth, sua patogênese é causada por uma anomalia hereditária dos nervos periféricos: motor (movimento) e sensorial (sensitivo).
Se o tipo de CMT for desmielinizante, a destruição ou defeito da bainha de mielina que protege os axônios dos nervos periféricos leva a uma desaceleração na transmissão dos impulsos nervosos no sistema nervoso periférico - entre o cérebro, os músculos e os órgãos sensoriais.
No tipo axonal da doença, os próprios axônios são afetados, o que afeta negativamente a força dos sinais nervosos, que é insuficiente para a estimulação completa dos músculos e órgãos sensoriais.
Leia também:
Como a síndrome de Charcot-Marie-Tooth é transmitida? Os genes defeituosos podem ser herdados de forma autossômica dominante ou autossômica recessiva.
O tipo mais comum, a herança autossômica dominante, ocorre quando há uma cópia do gene mutado (carregada por um dos pais). E a probabilidade de transmissão da CMT para cada filho nascido é estimada em 50%. [ 4 ]
Na herança autossômica recessiva, duas cópias do gene defeituoso (uma de cada pai que não apresenta nenhum sinal da doença) são necessárias para que a doença se desenvolva.
Em 40-50% dos casos ocorre desmielinização hereditária autossômica dominante, ou seja, CMT tipo 1; em 12-26% dos casos, CMT axonal, ou seja, tipo 2. E em 10-15% dos casos, observa-se herança ligada ao X. [ 5 ]
Sintomas Doença de Charcot-Marie-Tooth
Normalmente, os primeiros sinais desta doença começam a aparecer na infância e adolescência e se desenvolvem gradualmente ao longo da vida, embora a síndrome possa se manifestar mais tarde. A combinação de sintomas é variável e a taxa de progressão da doença, bem como sua gravidade, são impossíveis de prever.
Os sintomas típicos do estágio inicial incluem aumento da fadiga geral; diminuição do tônus (fraqueza) dos músculos dos pés, tornozelos e canelas; e ausência de reflexos. Isso dificulta a movimentação dos pés e leva à disbasia (distúrbio da marcha), que se manifesta pela elevação mais alta das pernas, frequentemente acompanhada de tropeços e quedas frequentes. Os sinais da doença de Charcot-Marie-Tooth em crianças pequenas podem incluir desajeitamento acentuado e dificuldades de caminhar, características da idade, associadas à queda bilateral do pé. Deformidades nos pés também são características: arco alto (pé cavo) ou pés chatos graves, dedos curvos (em forma de martelo).
No caso de andar na ponta dos pés em um contexto de hipotonia muscular, um neurologista pode suspeitar que a criança tenha CMT tipo 4, na qual as crianças podem ser incapazes de andar na adolescência.
À medida que a doença progride, a atrofia muscular e a fraqueza se espalham para as extremidades superiores, dificultando a coordenação motora fina e a execução de tarefas manuais normais. A diminuição das sensações táteis e da capacidade de sentir calor e frio, bem como dormência nos pés e nas mãos, indicam danos aos axônios dos nervos sensoriais.
Na doença de Charcot-Marie-Tooth tipos 3 e 6, que se manifesta na infância, são observados ataxia sensorial (coordenação prejudicada dos movimentos e equilíbrio), espasmos e tremores musculares, danos ao nervo facial, atrofia do nervo óptico com nistagmo e perda auditiva.
Em estágios posteriores, pode haver tremores incontroláveis e cãibras musculares frequentes; problemas de movimento podem levar ao desenvolvimento de dores: musculares, articulares e neuropáticas.
Complicações e consequências
A doença de Charcot-Marie-Tooth pode ter complicações e consequências como:
- entorses e fraturas mais frequentes;
- contraturas associadas ao encurtamento dos músculos e tendões periarticulares;
- escoliose (curvatura da coluna);
- problemas respiratórios – quando as fibras nervosas que inervam os músculos do diafragma são danificadas:
- perda da capacidade de se mover de forma independente.
Diagnósticos Doença de Charcot-Marie-Tooth
O diagnóstico inclui exame clínico, anamnese (incluindo histórico familiar), exame neurológico e sistêmico.
São realizados exames para verificar a amplitude de movimento, a sensibilidade e os reflexos tendinosos. A condução nervosa pode ser avaliada por meio de diagnósticos instrumentais – eletromiografia ou eletroneuromiografia. Ultrassonografia ou ressonância magnética também podem ser necessárias. [ 6 ]
Os testes genéticos ou de DNA para detectar as mutações genéticas mais comuns que causam CMT em uma amostra de sangue são limitados, pois os testes de DNA não estão disponíveis atualmente para todos os tipos da doença. Para mais informações, consulte Testes genéticos.
Em alguns casos, é realizada uma biópsia do nervo periférico (geralmente o nervo sural).
Diagnóstico diferencial
O diagnóstico diferencial inclui outras neuropatias periféricas, distrofia muscular de Duchenne, síndromes mielopáticas e miastênicas, neuropatia diabética, mielopatias na esclerose lateral múltipla e amiotrófica, síndrome de Guillain-Barré, trauma no nervo peroneal e sua atrofia (incluindo quando pinçado entre os discos lombares da coluna), danos ao cerebelo ou tálamo, bem como efeitos colaterais da quimioterapia (durante o tratamento com citostáticos como vincristina ou paclitaxel). [ 7 ]
Quem contactar?
Tratamento Doença de Charcot-Marie-Tooth
Atualmente, o tratamento para essa doença hereditária inclui terapia por exercícios (visando fortalecer e alongar os músculos); terapia ocupacional (que auxilia pacientes com fraqueza muscular nas mãos); e o uso de dispositivos ortopédicos para facilitar a marcha. Se necessário, analgésicos ou anticonvulsivantes são prescritos. [ 8 ]
Em casos de pés planos graves, pode ser realizada osteotomia e, em caso de deformação do calcanhar, está indicada a correção cirúrgica – artrodese. [ 9 ]
Pesquisas estão em andamento tanto sobre o componente genético da doença quanto sobre seus métodos de tratamento. O uso de células-tronco, certos hormônios, lecitina ou ácido ascórbico ainda não produziu resultados positivos.
Mas, graças a pesquisas recentes, algo novo pode de fato surgir no tratamento da doença de Charcot-Marie-Tooth em um futuro próximo. Assim, desde 2014, a empresa francesa Pharnext vem desenvolvendo, e desde meados de 2019, ensaios clínicos estão em andamento, o medicamento PXT3003 para o tratamento de CMT tipo 1 em adultos, que suprime o aumento da expressão do gene PMP22, melhora a mielinização dos nervos periféricos e alivia os sintomas neuromusculares.
Especialistas da empresa médica Sarepta Therapeutics (EUA) estão trabalhando na criação de uma terapia genética para a doença de Charcot-Marie-Tooth tipo 1. Essa terapia utilizará um vírus adenoassociado (AAV) inofensivo do gênero Dependovirus, com um genoma de DNA de fita simples linear, que transferirá o gene NTF3 para o corpo, codificando a proteína neurotrofina-3 (NT-3), necessária para o funcionamento das células nervosas de Schwann.
A Helixmith iniciará os ensaios clínicos da terapia genética desenvolvida na Coreia do Sul, Engensis (VM202), até o final de 2020, para tratar os sintomas musculares no CMT tipo 1. [ 10 ]
Prevenção
A prevenção da CMT pode ser feita por meio do aconselhamento genético de futuros pais, especialmente se alguém do casal tiver histórico familiar da doença. No entanto, já foram identificados casos de mutações genéticas pontuais de novo, ou seja, na ausência da doença no histórico familiar.
Durante a gravidez, a probabilidade da doença de Charcot-Marie-Tooth no futuro filho pode ser verificada por biópsia de vilo corial (de 10 a 13 semanas de gestação), bem como pela análise do líquido amniótico (de 15 a 18 semanas).
Previsão
Em geral, o prognóstico para os diferentes tipos de doença de Charcot-Marie-Tooth depende da gravidade clínica, mas em todos os casos a doença progride lentamente. Muitos pacientes apresentam deficiências, embora isso não reduza a expectativa de vida.