Médico especialista do artigo

Novas publicações
Nefrite hereditária (síndrome de Alport) em crianças
Última revisão: 05.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Nefrite hereditária (síndrome de Alport) é uma glomerulopatia hereditária não imune, determinada geneticamente, manifestada por hematúria (às vezes com proteinúria), declínio progressivo da função renal com desenvolvimento de insuficiência renal crônica, frequentemente combinada com surdez neurossensorial e deficiência visual.
A doença foi descrita pela primeira vez em 1902 por LG Guthrie, que observou uma família na qual a hematúria foi observada em várias gerações. Em 1915, AF Hurst descreveu o desenvolvimento de uremia em membros da mesma família. Em 1927, A. Alport identificou pela primeira vez perda auditiva em vários parentes com hematúria. Na década de 1950, lesões oculares em uma doença semelhante foram descritas. Em 1972, em pacientes com hematúria hereditária, durante um estudo morfológico do tecido renal, Hinglais et al. revelaram expansão e estratificação irregulares das membranas basais glomerulares. Em 1985, a base genética da nefrite hereditária foi identificada - uma mutação no gene do colágeno tipo IV (Fiengold et al., 1985).
O estudo da natureza genética da doença permitiu-nos concluir que as diferenças nas manifestações fenotípicas da nefrite hereditária (com ou sem perda auditiva) se devem ao grau de expressão do gene mutante. Assim, atualmente, todas as variantes clínicas são consideradas manifestações de uma única doença e o termo "nefrite hereditária" é sinônimo do termo "síndrome de Alport".
De acordo com estudos epidemiológicos, a nefrite hereditária ocorre com uma frequência de 17 por 100.000 crianças.
 [
[ Causas da Síndrome de Alport
A base genética da doença é uma mutação no gene da cadeia α-5 do colágeno tipo IV. Este tipo é universal para as membranas basais do rim, aparelho coclear, cápsula do cristalino, retina e córnea, o que foi comprovado em estudos utilizando anticorpos monoclonais contra essa fração do colágeno. Recentemente, foi indicada a possibilidade de utilização de sondas de DNA para o diagnóstico pré-natal de nefrite hereditária.
Ressalta-se a importância de testar todos os membros da família com sondas de DNA para identificar portadores do gene mutante, o que é de grande importância na condução do aconselhamento médico e genético de famílias com essa doença. No entanto, até 20% das famílias não têm parentes que sofrem de doença renal, o que sugere uma alta frequência de mutações espontâneas do gene anormal. A maioria dos pacientes com nefrite hereditária tem indivíduos com doença renal, perda auditiva e patologia da visão em suas famílias; casamentos consanguíneos entre pessoas com um ou mais ancestrais são importantes, uma vez que no casamento de indivíduos relacionados a probabilidade de receber os mesmos genes de ambos os pais aumenta. Rotas de transmissão autossômica dominante, autossômica recessiva e dominante ligadas ao cromossomo X foram estabelecidas.
Em crianças, três tipos de nefrite hereditária são mais comumente distinguidos: síndrome de Alport, nefrite hereditária sem perda auditiva e hematúria benigna familiar.
A síndrome de Alport é uma nefrite hereditária com deficiência auditiva. Baseia-se em um defeito combinado na estrutura do colágeno da membrana basal glomerular dos rins, ouvido e estruturas oculares. O gene da síndrome de Alport clássica está localizado no locus 21-22 q do braço longo do cromossomo X. Na maioria dos casos, é herdada de forma dominante, ligada ao cromossomo X. Nesse sentido, a síndrome de Alport é mais grave em homens, já que nas mulheres a função do gene mutante é compensada por um alelo saudável do segundo cromossomo não danificado.
A base genética para o desenvolvimento da nefrite hereditária são mutações nos genes das cadeias alfa do colágeno tipo IV. Seis cadeias alfa do colágeno tipo IV G são conhecidas: os genes das cadeias a5 e a6 (Col4A5 e Col4A5) estão localizados no braço longo do cromossomo X, na zona 21-22q; os genes das cadeias a3 e a4 (Col4A3 e Col4A4) estão no 2º cromossomo; os genes das cadeias a1 e a2 (Col4A1 e Col4A2) estão no 13º cromossomo.
Na maioria dos casos (80-85%), detecta-se um padrão de herança da doença ligado ao cromossomo X, associado a danos no gene Col4A5 como resultado de deleção, mutações pontuais ou distúrbios de splicing. Atualmente, foram encontradas mais de 200 mutações no gene Col4A5, responsáveis pela interrupção da síntese das cadeias α5 do colágeno tipo IV. Com esse tipo de herança, a doença se manifesta em crianças de ambos os sexos, mas é mais grave em meninos.
Mutações nos loci dos genes Col4A3 e Col4A4, responsáveis pela síntese das cadeias a3 e a4 do colágeno tipo IV, são herdadas de forma autossômica. Segundo pesquisas, a herança autossômica dominante é observada em 16% dos casos de nefrite hereditária, e a autossômica recessiva, em 6% dos pacientes. São conhecidas cerca de 10 variantes de mutações nos genes Col4A3 e Col4A4.
O resultado das mutações é uma violação dos processos de montagem do colágeno tipo IV, levando à violação de sua estrutura. O colágeno tipo IV é um dos principais componentes da membrana basal glomerular, do aparelho coclear e do cristalino, cuja patologia será detectada na clínica da nefrite hereditária.
O colágeno tipo IV, que faz parte da membrana basal glomerular, consiste principalmente em duas cadeias a1 (IV) e uma cadeia a2 (IV), e também contém cadeias a3, a4 e a5. Na maioria das vezes, na herança ligada ao cromossomo X, a mutação do gene Col4A5 é acompanhada pela ausência das cadeias a3, a4, a5 e a6 na estrutura do colágeno tipo IV, e o número de cadeias o1 e a2 na membrana basal glomerular aumenta. O mecanismo desse fenômeno não é claro; presume-se que a causa sejam alterações pós-transcricionais no mRNA.
A ausência das cadeias a3, a4 e a5 na estrutura do colágeno tipo IV das membranas basais glomerulares leva ao seu adelgaçamento e fragilidade nos estágios iniciais da síndrome de Alport, que se manifesta clinicamente mais frequentemente por hematúria (menos frequentemente por hematúria com proteinúria ou apenas proteinúria), perda auditiva e lenticone. A progressão da doença leva ao espessamento e à diminuição da permeabilidade das membranas basais nos estágios finais da doença, com proliferação de colágenos tipos V e VI, manifestada por aumento da proteinúria e diminuição da função renal.
A natureza da mutação subjacente à nefrite hereditária determina em grande parte sua manifestação fenotípica. No caso de deleção do cromossomo X com mutação simultânea dos genes Col4A5 e Col4A6, responsáveis pela síntese das cadeias a5 e a6 do colágeno tipo IV, a síndrome de Alport é combinada com leiomiomatose do esôfago e dos genitais. De acordo com dados de pesquisa, no caso de uma mutação do gene Col4A5 associada a uma deleção, observa-se uma maior gravidade do processo patológico, uma combinação de dano renal com manifestações extrarrenais e desenvolvimento precoce de insuficiência renal crônica, em comparação com uma mutação pontual desse gene.
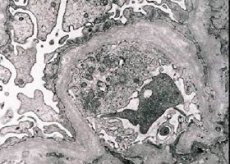
Morfologicamente, a microscopia eletrônica revela adelgaçamento e estratificação das membranas basais glomerulares (especialmente da lâmina densa) e a presença de grânulos eletrondensos. As lesões glomerulares podem ser heterogêneas no mesmo paciente, desde lesões mesangiais focais mínimas até glomeruloesclerose. A glomerulite na síndrome de Alport é sempre imunonegativa, o que a distingue da glomerulonefrite. Os sinais característicos incluem o desenvolvimento de atrofia tubular, infiltração linfo-histiocitária e a presença de "células espumosas" com inclusões lipídicas - lipófagos. À medida que a doença progride, revelam-se espessamento e destruição pronunciada das membranas basais glomerulares.
Certas alterações no sistema imunológico são reveladas. Pacientes com nefrite hereditária apresentam níveis reduzidos de IgA e tendência a aumentar a concentração de IgM no sangue. Os níveis de IgG podem estar aumentados nos estágios iniciais da doença e diminuir nos estágios mais avançados. Talvez o aumento da concentração de IgM e G seja uma espécie de reação compensatória em resposta à deficiência de IgA.
A atividade funcional do sistema de linfócitos T é reduzida; observa-se uma diminuição seletiva dos linfócitos B responsáveis pela síntese de Ig A, a ligação fagocítica da imunidade é interrompida, principalmente devido à interrupção dos processos de quimiotaxia e digestão intracelular em neutrófilos
Ao examinar uma biópsia renal em pacientes com síndrome de Alport, os dados da microscopia eletrônica revelam alterações ultraestruturais na membrana basal glomerular: afinamento, ruptura da estrutura e divisão das membranas basais glomerulares com alteração em sua espessura e contornos irregulares. Nos estágios iniciais da nefrite hereditária, o defeito determina o afinamento e a fragilidade das membranas basais glomerulares.
O afinamento das membranas glomerulares é um sinal mais favorável e mais comum em meninas. Um sinal microscópico eletrônico mais constante na nefrite hereditária é a ruptura da membrana basal, e a gravidade de sua destruição se correlaciona com a gravidade do processo.
Sintomas da Síndrome de Alport em Crianças
Os primeiros sintomas da síndrome de Alport, na forma de síndrome urinária isolada, são mais frequentemente detectados em crianças nos primeiros três anos de vida. Na maioria dos casos, a doença é detectada por acaso. A síndrome urinária é detectada durante um exame preventivo da criança, antes da admissão em uma creche ou durante a SARS. Em caso de patologia na urina durante a SARS. Na nefrite hereditária, ao contrário da glomerulonefrite adquirida, não há período latente.
No estágio inicial da doença, a saúde da criança sofre pouco, sendo a persistência e a resistência da síndrome urinária uma característica marcante. Um dos principais sinais é a hematúria, de gravidade variável, observada em 100% dos casos. Um aumento no grau de hematúria é observado durante ou após infecções respiratórias, atividade física ou após vacinações preventivas. A proteinúria, na maioria dos casos, não ultrapassa 1 g/dia; no início da doença, pode ser instável; à medida que o processo progride, a proteinúria aumenta. Periodicamente, pode haver leucocitúria com predomínio de linfócitos no sedimento urinário, o que está associado ao desenvolvimento de alterações intersticiais.
Posteriormente, a função renal parcial é prejudicada e o estado geral do paciente piora: intoxicação, fraqueza muscular, hipotensão arterial, frequentemente deficiência auditiva (especialmente em meninos) e, às vezes, deficiência visual. A intoxicação se manifesta por palidez, fadiga e dores de cabeça. No estágio inicial da doença, a perda auditiva é, na maioria dos casos, detectada apenas pela audiografia. A perda auditiva na síndrome de Alport pode ocorrer em diferentes períodos da infância, mas na maioria das vezes a perda auditiva é diagnosticada entre 6 e 10 anos de idade. A perda auditiva em crianças começa nas altas frequências, atingindo um grau significativo na condução aérea e óssea, passando da perda auditiva condutora para a perceptiva. A perda auditiva pode ser um dos primeiros sintomas da doença e pode preceder a síndrome urinária.
Em 20% dos casos, pacientes com síndrome de Alport apresentam alterações nos órgãos visuais. As anomalias mais frequentemente detectadas são as do cristalino: esferofoquia, lenticono anterior, posterior ou misto e várias cataratas. Em famílias com síndrome de Alport, há uma frequência significativa de miopia. Vários pesquisadores observam constantemente alterações perimaculares bilaterais nessas famílias na forma de granulações esbranquiçadas ou amareladas brilhantes no corpo lúteo. Eles consideram esse sinal um sintoma constante com alto valor diagnóstico na síndrome de Alport. KS Chugh et al. (1993) em um estudo oftalmológico encontraram em pacientes com síndrome de Alport uma diminuição da acuidade visual em 66,7% dos casos, lenticono anterior em 37,8%, manchas retinianas em 22,2%, catarata em 20% e ceratocone em 6,7%.
Em algumas crianças com nefrite hereditária, especialmente quando se desenvolve insuficiência renal, observa-se um atraso significativo no desenvolvimento físico. À medida que a insuficiência renal progride, desenvolve-se hipertensão arterial. Em crianças, é mais frequentemente detectada na adolescência e em faixas etárias mais avançadas.
Pacientes com nefrite hereditária são caracterizados pela presença de vários estigmas (mais de 5 a 7) de dismorfogênese do tecido conjuntivo. Entre os estigmas do tecido conjuntivo em pacientes, os mais comuns são hipertelorismo ocular, palato ogival, anomalias de mordida, formato anormal das aurículas, curvatura do dedo mínimo nas mãos e "espaço de sandália" nos pés. A nefrite hereditária é caracterizada pela uniformidade dos estigmas de dismorfogênese dentro de uma família, bem como pela alta frequência de sua distribuição entre parentes de probandos por cuja linhagem a doença é transmitida.
Nos estágios iniciais da doença, detecta-se uma diminuição isolada das funções renais parciais: transporte de aminoácidos, eletrólitos, função de concentração, acidogênese. Alterações posteriores afetam o estado funcional das partes proximal e distal do néfron e são caracterizadas por distúrbios parciais combinados. Uma diminuição da filtração glomerular ocorre mais tarde, mais frequentemente na adolescência. À medida que a nefrite hereditária progride, desenvolve-se anemia.
Assim, a nefrite hereditária é caracterizada por um curso da doença em estágios: primeiro, um estágio latente ou sintomas clínicos ocultos, manifestados por alterações mínimas na síndrome urinária, seguido de uma descompensação gradual do processo com diminuição da função renal e sintomas clínicos manifestos (intoxicação, astenia, atraso no desenvolvimento, anemia). Os sintomas clínicos geralmente aparecem independentemente da estratificação da reação inflamatória.
A nefrite hereditária pode se manifestar em diferentes períodos etários, o que depende da ação do gene, que fica em estado reprimido até certo momento.
Classificação
Existem três tipos de nefrite hereditária
- Opção I - manifesta-se clinicamente como nefrite com hematúria, perda auditiva e lesão ocular. A evolução da nefrite é progressiva com o desenvolvimento de insuficiência renal crônica. O tipo de herança é dominante, ligado ao cromossomo X. Morfologicamente, revela-se uma violação da estrutura da membrana basal, seu afinamento e divisão.
- Opção II - manifesta-se clinicamente como nefrite com hematúria sem perda auditiva. A evolução da nefrite é progressiva com o desenvolvimento de insuficiência renal crônica. O tipo de herança é dominante, ligado ao cromossomo X. Morfologicamente, detecta-se afinamento da membrana basal dos capilares glomerulares (especialmente da lâmina densa).
- Opção III - hematúria familiar benigna. A evolução é favorável, sem desenvolvimento de insuficiência renal crônica. O tipo de herança é autossômico dominante ou autossômico recessivo. Na herança autossômica recessiva, observa-se um curso mais grave da doença em mulheres.
Diagnóstico da síndrome de Alport
Os seguintes critérios são propostos:
- a presença de pelo menos dois pacientes com nefropatia em cada família;
- hematúria como principal sintoma de nefropatia no probando;
- a presença de perda auditiva em pelo menos um membro da família;
- desenvolvimento de insuficiência renal crônica em um ou mais parentes.
No diagnóstico de diversas doenças hereditárias e congênitas, destaca-se a importância de uma abordagem abrangente do exame e, acima de tudo, a atenção aos dados obtidos na compilação do heredograma da criança. O diagnóstico da síndrome de Alport é considerado válido nos casos em que 3 dos 4 sinais típicos são detectados no paciente: presença de hematúria e insuficiência renal crônica na família, presença de perda auditiva neurossensorial, patologia visual no paciente e detecção de sinais de clivagem da membrana basal glomerular com alteração em sua espessura e contornos irregulares durante as características microscópicas eletrônicas da biópsia.
O exame do paciente deve incluir métodos de pesquisa clínica e genética; estudo direcionado do histórico da doença; exame geral do paciente, levando em consideração critérios diagnósticos significativos. No estágio de compensação, a patologia só pode ser detectada com foco em síndromes como a presença de carga hereditária, hipotensão, múltiplos estigmas de disembriogênese e alterações na síndrome urinária. No estágio de descompensação, podem surgir sintomas extrarrenais, como intoxicação grave, astenia, atraso no desenvolvimento físico, anemia, que se manifestam e se intensificam com uma diminuição gradual da função renal. Na maioria dos pacientes, com diminuição da função renal, observa-se o seguinte: diminuição da acidogênese e da aminogênese; 50% dos pacientes notam uma diminuição significativa da função secretora dos rins; amplitude limitada de flutuações na densidade óptica da urina; distúrbio do ritmo de filtração e, em seguida, diminuição da filtração glomerular. O estágio da insuficiência renal crônica é diagnosticado quando os pacientes apresentam um nível elevado de ureia no soro sanguíneo (mais de 0,35 g/l) por 3 a 6 meses ou mais, e uma diminuição na filtração glomerular para 25% do normal.
O diagnóstico diferencial da nefrite hereditária deve ser realizado principalmente com a forma hematúrica da glomerulonefrite adquirida. A glomerulonefrite adquirida geralmente tem início agudo, um período de 2 a 3 semanas após a infecção, sinais extrarrenais, incluindo hipertensão desde os primeiros dias (na nefrite hereditária, ao contrário, hipotensão), diminuição da filtração glomerular no início da doença, sem comprometimento das funções tubulares parciais, enquanto na hereditária elas estão presentes. A glomerulonefrite adquirida cursa com hematúria e proteinúria mais pronunciadas, com aumento da VHS. Alterações típicas na membrana basal glomerular, características da nefrite hereditária, são de valor diagnóstico.
O diagnóstico diferencial da nefropatia dismetabólica é realizado com insuficiência renal crônica, com doenças renais heterogêneas clinicamente demonstradas na família, podendo haver um espectro de nefropatia que vai da pielonefrite à urolitíase. As crianças frequentemente apresentam queixas de dor abdominal e, ocasionalmente, ao urinar, com presença de oxalatos no sedimento urinário.
Se houver suspeita de nefrite hereditária, o paciente deve ser encaminhado a um serviço de nefrologia especializado para esclarecer o diagnóstico.
O que precisa examinar?
Como examinar?
Quais testes são necessários?
Quem contactar?
Tratamento da síndrome de Alport
O regime inclui restrições a esforços físicos intensos e exposição ao ar livre. A dieta é completa, com níveis suficientes de proteínas, gorduras e carboidratos completos, levando em consideração a função renal. De grande importância é a detecção e o tratamento de focos crônicos de infecção. Os seguintes medicamentos são utilizados: ATP, cocarboxilase, piridoxina (até 50 mg/dia), cloreto de carnitina. Os tratamentos são realizados de 2 a 3 vezes por ano. Para hematúria, são prescritos fitoterápicos - urtiga, suco de chokeberry, mil-folhas.
Há relatos na literatura nacional e estrangeira sobre o tratamento com prednisolona e o uso de citostáticos. No entanto, é difícil avaliar o efeito.
Na insuficiência renal crônica, são utilizados hemodiálise e transplante renal.
Não existem métodos de terapia patogênica específica (eficaz) para nefrite hereditária. Todas as medidas de tratamento visam prevenir e retardar o declínio da função renal.
A dieta deve ser balanceada e hipercalórica, levando em consideração o estado funcional dos rins. Na ausência de distúrbios funcionais, a dieta da criança deve conter proteínas, gorduras e carboidratos suficientes. Na presença de sinais de disfunção renal, a quantidade de proteínas, carboidratos, cálcio e fósforo deve ser limitada, o que retarda o desenvolvimento de insuficiência renal crônica.
A atividade física deve ser limitada; é aconselhável que as crianças evitem esportes.
O contato com pacientes infecciosos deve ser evitado, reduzindo o risco de desenvolvimento de doenças respiratórias agudas. A higienização de focos de infecção crônica é necessária. Vacinações preventivas não são realizadas para crianças com nefrite hereditária; a vacinação só é possível por motivos epidemiológicos.
A terapia hormonal e imunossupressora para nefrite hereditária é ineficaz. Há indícios de algum efeito positivo (redução da proteinúria e desaceleração da progressão da doença) com o uso prolongado de ciclosporina A e inibidores da ECA por vários anos.
No tratamento de pacientes são utilizados medicamentos que melhoram o metabolismo:
- piridoxina - 2-3 mg/kg/dia em 3 doses por 4 semanas;
- cocarboxilase - 50 mg por via intramuscular em dias alternados, totalizando 10-15 injeções;
- ATP - 1 ml por via intramuscular em dias alternados, 10-15 injeções;
- vitamina A - 1000 UI/ano/dia em 1 dose por 2 semanas;
- Vitamina E - 1 mg/kg/dia em 1 dose por 2 semanas.
Este tipo de terapia ajuda a melhorar o estado geral dos pacientes, reduzir disfunções tubulares e é realizado em cursos 3 vezes ao ano.
O levamisol pode ser usado como imunomodulador - 2 mg/kg/dia 2 a 3 vezes por semana com intervalos entre as doses de 3 a 4 dias.
De acordo com dados de pesquisa, a oxigenação hiperbárica tem um efeito positivo na gravidade da hematúria e da disfunção renal.
O método mais eficaz para tratar a nefrite hereditária é o transplante renal oportuno. Nesse caso, não há recidiva da doença no transplante; em uma pequena porcentagem de casos (cerca de 5%), a nefrite pode se desenvolver no rim transplantado, associada a antígenos da membrana basal glomerular.
Uma direção promissora é o diagnóstico pré-natal e a terapia de engenharia genética. Experimentos em animais demonstram alta eficiência na transferência de genes normais responsáveis pela síntese de cadeias alfa de colágeno tipo IV para o tecido renal, após o que se observa a síntese de estruturas normais de colágeno.
Previsão
O prognóstico da nefrite hereditária é sempre sério.
Os critérios prognósticos desfavoráveis para o curso da nefrite hereditária são:
- gênero masculino;
- desenvolvimento precoce de insuficiência renal crônica em familiares;
- proteinúria (mais de 1 g/dia);
- espessamento das membranas basais glomerulares segundo microscopia;
- neurite acústica;
- deleção no gene Col4A5.
O prognóstico para hematúria familiar benigna é mais favorável.
Использованная литература