Médico especialista do artigo

Novas publicações
Queratodermia: causas, sintomas, diagnóstico, tratamento
Última revisão: 07.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
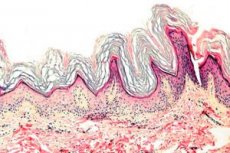
Ceratodermia é um grupo de dermatoses caracterizadas por uma interrupção do processo de queratinização - formação excessiva de células córneas principalmente nas palmas das mãos e plantas dos pés.
As causas e a patogênese da doença não foram totalmente esclarecidas. Pesquisas estabeleceram que as ceratodermias são causadas por mutações nos genes que codificam a queratina 6, 9 e 16. Deficiência de vitamina A, disfunções hormonais, principalmente das glândulas sexuais, e infecções bacterianas e virais são de grande importância na patogênese. São um dos sintomas de doenças hereditárias e tumores de órgãos internos (ceratodermias parapsoriáticas).
Sintomas. Distingue-se entre ceratodermia difusa (ceratodermia de Unna-Tost, ceratodermia de Meleda, ceratodermia de Papillon-Lefèvre, ceratodermia mutilante e síndromes que incluem ceratodermia difusa como um dos principais sintomas) e focal (ceratodermia disseminada de Fischer-Buschke, acroceratoelastoidose de Kosti, ceratodermia limitada de Bruhauer-Franzesthesti, ceratodermia linear de Fuchs, etc.).
A ceratodermia de Winy-Tost (sinônimos: ictiose congênita das palmas e plantas dos pés, síndrome de Winy-Tost) é transmitida de forma autossômica dominante. Há uma queratinização excessiva difusa da pele das palmas das mãos e plantas dos pés (às vezes apenas das plantas dos pés), que se desenvolve nos primeiros dois anos de vida. O processo patológico da pele inicia-se com um ligeiro espessamento da pele das palmas das mãos e plantas dos pés na forma de uma faixa de eritema de cor lívida na borda com a pele sã. Com o tempo, camadas córneas lisas e amareladas aparecem em sua superfície. A lesão raramente se espalha para o dorso dos punhos ou dedos. Em alguns pacientes, podem formar-se fissuras superficiais ou profundas e observa-se hiperidrose local. No paciente observado pelo autor, o tio materno, o irmão e o filho sofriam de ceratodermia de Winy-Tost.
São descritos casos de danos às unhas (espessamento), dentes e cabelos na ceratodermia de Winy-Tost em combinação com várias anomalias esqueléticas e patologias de órgãos internos, sistemas nervoso e endócrino.
Histopatologia. O exame histológico revela hiperceratose acentuada, granulose, acantose e pequenos infiltrados inflamatórios na derme superior. Diagnóstico diferencial. A doença deve ser diferenciada de outros tipos de ceratodermia.
A ceratodermia de Meleda (sinônimos: doença de Meleda, acroceratoma progressivo congênito, ceratose palmoplantar transgradiente de Siemens, ceratose palmoplantar progressiva hereditária de Kogoy) é herdada de forma autossômica recessiva. Essa forma de ceratodermia é caracterizada por espessas camadas córneas amarelo-acastanhadas com fissuras profundas. Uma borda violeta-púrpura com vários milímetros de largura é visível ao longo das bordas da lesão. O processo geralmente se espalha para o dorso das mãos e pés, antebraços e canelas. A maioria dos pacientes apresenta hiperidrose local. Nesse caso, a superfície das palmas e plantas dos pés fica levemente úmida e coberta por pontos pretos (ductos das glândulas sudoríparas).
A doença pode se desenvolver por volta dos 15-20 anos de idade. As unhas engrossam e ficam deformadas.
Histopatologia. O exame histológico revela hiperceratose, às vezes acantose, e um infiltrado inflamatório crônico na derme papilar.
Diagnóstico diferencial. A ceratodermia de Melela deve ser diferenciada da ceratodermia de Unna-Tost.
Ceratodermia Papillon-Lefevre (sinônimo: hiperceratose palmoplantar com periodontite) é herdada de forma autossômica recessiva.
A doença se manifesta entre o 2º e o 3º ano de vida. O quadro clínico da doença é semelhante ao da doença de Melela. Além disso, as alterações nos dentes são características (anormalidades na erupção dos dentes de leite e permanentes com desenvolvimento de cáries, gengivite, periodontite de progressão rápida com perda prematura dos dentes).
Histopatologia. O exame histológico revela espessamento de todas as camadas da epiderme, especialmente da camada córnea, e aglomerados celulares insignificantes de linfócitos e histiócitos na derme.
Diagnóstico diferencial. A doença deve ser diferenciada de outras ceratodermias. Uma importante característica distintiva é a patologia dentária característica, que não é encontrada em outras formas de ceratodermia difusa hereditária.
Ceratodermia mutilante (sinônimos: síndrome de Fonwinkel, ceratoma mutilante hereditário) é um tipo de ceratodermia difusa, herdada de forma autossômica dominante. Desenvolve-se no segundo ano de vida e caracteriza-se por depósitos córneos difusos na pele das palmas das mãos e plantas dos pés, com hiperidrose. Com o tempo, formam-se sulcos em forma de cordão nos dedos, o que leva a contraturas e amputação espontânea dos dedos. A ceratose folicular manifesta-se no dorso das mãos, bem como na região das articulações do cotovelo e do joelho. As lâminas ungueais são alteradas (frequentemente como vidros de relógio). Foram descritos casos de hipogonadismo, alopecia rubi, perda auditiva e paquioníquia.
Histopatologia. O exame histológico revela hiperceratose grave, granulose, acantose e pequenos infiltrados inflamatórios na derme, constituídos por linfócitos e histiócitos.
Diagnóstico diferencial. Ao diferenciar a ceratodermia mutilante de outras formas de ceratodermia difusa, o efeito da mutilação, que não é típico de outras formas, deve ser levado em consideração em primeiro lugar. Ao realizar o diagnóstico diferencial de todas as formas de ceratodermia difusa, é necessário lembrar que ela pode ser um dos principais sintomas de diversas síndromes hereditárias.
Tratamento. A neotigazona é indicada no tratamento geral da ceratodermia. A dose do medicamento depende da gravidade do processo e é de 0,3 a 1 mg/kg de peso corporal do paciente. Na ausência de neotigazona, recomenda-se vitamina A na dose de 100 a 300.000 mg por dia, por um período prolongado. O tratamento externo consiste no uso de pomadas com retinoides aromáticos, agentes ceratolíticos e esteroides.
 [
[ O que está incomodando você?
O que precisa examinar?
Como examinar?