Médico especialista do artigo

Novas publicações
Linfomas de células T da pele
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
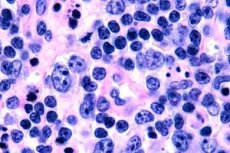
Os linfomas de células T são mais frequentemente registrados em idosos, embora casos isolados da doença sejam observados até mesmo em crianças. Os homens adoecem duas vezes mais que as mulheres. Os linfomas de células T são de natureza epidermotrópica.
Causas Linfomas de células T da pele
As causas e a patogênese dos linfomas cutâneos de células T não são totalmente compreendidas. Atualmente, a maioria dos pesquisadores considera o vírus da leucemia de células T humanas tipo 1 (HTLV-1) I como o principal fator etiológico que inicia o desenvolvimento de linfomas malignos de células T da pele. Além disso, discute-se o papel de outros vírus no desenvolvimento do linfoma de células T: vírus Epstein-Barr e herpes simplex tipo 6. Em pacientes com linfoma de células T, os vírus são encontrados na pele, no sangue periférico e nas células de Langerhans. Anticorpos contra o HTLV-I são detectados em muitos pacientes com micose fungoide.
Um lugar importante na patogênese dos linfomas de células T é desempenhado por processos imunopatológicos na pele, sendo o principal a proliferação descontrolada de linfócitos clonais.
As citocinas produzidas por linfócitos, células epiteliais e células do sistema macrofágico têm efeitos pró-inflamatórios e proliferativos (IL-1, responsável pela diferenciação dos linfócitos; IL-2 - fator de crescimento de células T; IL-4 e IL-5, que aumentam o influxo de eosinófilos na lesão e sua ativação, etc.). Como resultado do influxo de linfócitos T na lesão, formam-se microabscessos de Pautrier. Simultaneamente ao aumento da proliferação de linfócitos, a atividade das células de defesa antitumorais é suprimida: células assassinas naturais, linfócitos linfocitotóxicos, células dendríticas, em particular, células de Langerhans, bem como citocinas (IL-7, IL-15, etc.) - inibidores do crescimento tumoral. O papel de fatores hereditários não pode ser descartado. A presença de casos familiares e a detecção frequente de alguns antígenos de histocompatibilidade (HLA B-5 e HLA B-35 - em linfomas de pele altamente malignos, HLA A-10 - em linfomas menos agressivos, HLA B-8 - na forma eritrodérmica da micose fungoide) confirmam a natureza hereditária da dermatose.
Observações clínicas indicam uma possível transformação de dermatoses crônicas de longa duração (neurodermatite, dermatite atópica, psoríase, etc.) em micose fungoide. O fator-chave é a persistência prolongada de linfócitos no foco inflamatório, o que interrompe a vigilância imunológica e promove o surgimento de um clone de linfócitos malignos e, consequentemente, o desenvolvimento de um processo proliferativo maligno.
O impacto de fatores físicos no corpo, como insolação, radiação ionizante e substâncias químicas, pode levar ao surgimento de um clone de linfócitos “genotraumáticos” que têm efeito mutagênico nas células linfoides e ao desenvolvimento de malignidade linfocitária.
Portanto, os linfomas de células T podem ser considerados uma doença multifatorial que se inicia com a ativação de linfócitos sob a influência de diversos fatores carcinogênicos e "genotraumatizantes" e com o surgimento de um clone dominante de células T. A gravidade do distúrbio de vigilância imunológica, o clone de linfócitos malignos, determina as manifestações clínicas (elementos maculosos, em placas ou tumorais) dos linfomas de células T.
Patogênese
No estágio inicial da micose fungoide, observam-se acantose com processos amplos, hiperplasia e compactação dos queratinócitos basais, degeneração vacuolar de algumas células basais, mitoses atípicas em diferentes camadas da epiderme e epidermotropismo do infiltrado com penetração de linfócitos na epiderme. Na derme, observam-se pequenos infiltrados ao redor dos vasos, constituídos por células mononucleares únicas com núcleos hipercrômicos - células "micóticas". No segundo estágio, observa-se um aumento na gravidade do infiltrado dérmico e epidermotropismo das células infiltradas, resultando na penetração de linfócitos malignos na epiderme, formando aglomerados na forma de microabscessos de Potrier. No terceiro estágio, tumoral, observam-se acantose maciça e leve atrofia da epiderme, além de aumento da infiltração da epiderme por linfócitos tumorais, que formam múltiplos microabscessos de Potrier. O infiltrado maciço está localizado em toda a espessura da derme e cobre parte da hipoderme. Observam-se formas blásticas de linfócitos.
Linfoma anaplásico cutâneo de grandes células T
É representado por um grupo de processos linfoproliferativos caracterizados pela presença de proliferações de células T CD30+ anaplásicas clonais atípicas. Via de regra, desenvolve-se secundariamente na fase tumoral da micose fungoide ou na síndrome de Sézary, mas pode desenvolver-se de forma independente ou com disseminação de linfomas sistêmicos desse tipo. Clinicamente, esses linfomas correspondem à chamada forma decapitada da micose fungoide, apresentando-se como linfonodos únicos ou múltiplos, geralmente agrupados.
Histologicamente, o proliferado ocupa quase toda a derme com ou sem epidermotropismo no caso de atrofia epidérmica.
Citologicamente, as células tumorais podem variar em tamanho e forma. Com base nessas propriedades, distingue-se entre linfoma pleomórfico de células T de células médias e grandes, com núcleos de várias configurações irregulares – convoluto, multilobado, com cromatina densa, nucléolo bem definido e citoplasma bastante abundante; imunoblástico – com núcleos grandes, redondos ou ovais, com carioplasma claro e um nucléolo central; anaplásico – com células muito grandes e feias, com núcleos de configuração irregular e citoplasma abundante. Fenotipicamente, todo esse grupo pertence aos linfomas T-helper e pode ser CD30+ ou CD30-.
R. Willemze et al. (1994) demonstraram que o curso do linfoma CD30+ é mais favorável. Genotipicamente, detecta-se rearranjo clonal do receptor de linfócitos T.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Sintomas Linfomas de células T da pele
A doença mais comum no grupo dos linfomas de células T da pele é a micose fungoide, responsável por cerca de 70% dos casos. Existem três formas clínicas da doença: clássica, eritrodérmica e decapitada. Os linfomas de células T são caracterizados pelo polimorfismo de erupções cutâneas na forma de manchas, placas e tumores.
A forma eritrodérmica da micose fungoide geralmente começa com prurido incontrolável, edema, hiperemia generalizada e aparecimento de lesões eritemato-escamosas na pele do tronco e das extremidades, que tendem a se fundir e desenvolver eritrodermia em 1 a 2 meses. Quase todos os pacientes apresentam hiperceratose palmo-plantar e afinamento difuso dos pelos por toda a pele. Todos os grupos de linfonodos estão bastante aumentados. Os linfonodos inguinais, femorais, axilares e cubitais aumentados são palpados como "pacotes" de consistência elástica densa, não fundidos com os tecidos circundantes e indolores. O estado geral piora acentuadamente: ocorrem febre com temperatura corporal de até 38-39 °C, suores noturnos, fraqueza e perda de peso. Atualmente, a síndrome de Sézary é considerada por muitos dermatologistas como a variante leucêmica mais rara da forma eritrodérmica da micose fungoide.
Linfocitogramas mostram leucocitose pronunciada - células de Sézary. As células de Sézary são células T auxiliares malignas, cujos núcleos apresentam uma superfície cerebriforme dobrada com invaginações profundas da membrana nuclear. Observa-se um desfecho fatal após 2 a 5 anos, cuja causa frequente é patologia cardiovascular e intoxicação.
A forma decapitada da micose fungoide é caracterizada pelo rápido desenvolvimento de lesões tumorais em pele aparentemente saudável, sem formação prévia de placa a longo prazo. Esta forma é caracterizada por um alto grau de malignidade, sendo considerada uma manifestação de linfossarcoma. A morte é observada em até um ano.
Estágios
A forma clássica da micose fungoide é caracterizada por três estágios de desenvolvimento: eritemato-escamoso, em placas e tumoral.
O primeiro estágio assemelha-se ao quadro clínico de algumas dermatoses inflamatórias benignas - eczema, dermatite seborreica, parapsoríase em placas. Nesta fase da doença, observam-se manchas de vários tamanhos, intensamente rosadas, rosa-avermelhadas com tonalidade púrpura, de contornos redondos ou ovais, com limites relativamente nítidos, descamação superficial em forma de farelo ou em placas finas. Os elementos frequentemente se localizam em diferentes áreas da pele, mais frequentemente no tronco e na face. Gradualmente, seu número aumenta. Com o tempo, o processo pode assumir o caráter de eritrodermia (estágio eritrodérmico). A erupção cutânea pode persistir por anos ou desaparecer espontaneamente. Ao contrário das dermatoses inflamatórias benignas, os elementos da erupção cutânea e da coceira nesta fase são resistentes à terapia.
A fase infiltrativa em placas desenvolve-se ao longo de vários anos. No lugar de erupções cutâneas previamente existentes, surgem placas de contornos arredondados ou irregulares, de coloração intensamente roxa, claramente delimitadas da pele saudável, densas e com superfície escamosa. Sua consistência assemelha-se a "papelão grosso". Algumas delas se resolvem espontaneamente, deixando áreas de hiperpigmentação marrom-escura e/ou atrofia (poiquilodermia). A coceira nesta fase é ainda mais intensa e dolorosa, com febre e perda de peso. Linfadenopatia pode ser observada nesta fase.
No terceiro estágio, o tumor, surgem tumores indolores, de consistência densa e elástica, de coloração amarelo-avermelhada, que se desenvolvem a partir de placas ou surgem em pele aparentemente saudável. O formato dos tumores é esférico ou achatado, frequentemente assemelhando-se a um cogumelo. Os tumores podem aparecer em qualquer lugar. Seu número varia amplamente, de um a dezenas, e seu tamanho varia de 1 a 20 cm de diâmetro. Quando tumores de longa data se desintegram, formam-se úlceras com bordas irregulares e fundo profundo, que atingem a fáscia ou o osso. Os linfonodos, o baço, o fígado e os pulmões são os mais frequentemente afetados. O estado geral piora, os sintomas de intoxicação aparecem e aumentam e a fraqueza se desenvolve. A expectativa de vida média dos pacientes com a forma clássica de micose fungoide, desde o momento do diagnóstico, é de 5 a 10 anos. A mortalidade geralmente é observada por doenças intercorrentes: pneumonia, insuficiência cardiovascular, amiloidose. Subjetivamente, sente-se coceira e, quando os tumores se desintegram, dor nas áreas afetadas.
O que precisa examinar?
Como examinar?
Tratamento Linfomas de células T da pele
No estágio eritemato-escamoso, os pacientes não necessitam de terapia antitumoral; são prescritos corticosteroides tópicos (prednisolona, betametasona, derivados de dexametasona), interferon alfa (3 milhões de UI por dia, seguidos de 3 vezes por semana durante 3 a 6 meses, dependendo das manifestações clínicas ou da eficácia do tratamento), interferon gama (100.000 UI por dia durante 10 dias, repetindo o ciclo de 12 a 3 vezes com intervalo de 10 dias), terapia PUVA ou terapia Re-PUVA. A eficácia da terapia PUVA baseia-se na formação seletiva de ligações cruzadas covalentes dos psoralenos com o DNA nas células T auxiliares em proliferação, o que inibe sua divisão. Na segunda fase, além dos agentes mencionados, são utilizados corticosteroides sistêmicos (30-40 mg de prednisolona por dia durante 1,5 a 2 meses) e citostáticos (prospedina 100 mg por dia, 4 a 5 injeções no total). A combinação de interferons com outros métodos terapêuticos tem um efeito terapêutico mais pronunciado (interferons + PUVA, interferons + citostáticos, interferons + retinoides aromáticos).
No estágio tumoral, o principal método é a poliquimioterapia. Utiliza-se uma combinação de vincristina (0,5-1 mg por via intravenosa uma vez ao dia, totalizando 4-5 injeções) com prednisolona (40-60 mg por dia por via oral durante a quimioterapia), prospidina (100 mg por dia, totalizando 3 g) e interferons. Recomenda-se fototerapia, terapia por feixe de elétrons e fotoférese (fotoquimioterapia extracorpórea).