Médico especialista do artigo

Novas publicações
Síndrome de Treacher Collins
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.

Distúrbios intrauterinos nos processos de desenvolvimento ósseo causam graves deformidades craniofaciais, e uma das variedades dessa patologia é a síndrome de Treacher Collins (STC) ou mandibulofascial, ou seja, disostose maxilofacial.
Código da doença segundo CID 10: classe XVII (anomalias congênitas, deformidades e distúrbios cromossômicos), Q75.4 - disostose mandibulofacial.
Causas Síndrome de Treacher Collins
Esta síndrome recebeu o nome em homenagem ao destacado oftalmologista britânico Edward Treacher Collins, que descreveu as principais características da patologia há mais de cem anos. No entanto, os médicos europeus costumam chamar esse tipo de anomalia facial e óssea da mandíbula de doença ou síndrome de Franceschetti – com base na extensa pesquisa do oftalmologista suíço Adolf Franceschetti, que introduziu o termo "disostose mandibulofascial" em meados do século passado. No meio médico, o termo "síndrome de Franceschetti-Collins" também é utilizado.
A síndrome de Treacher Collins é causada por mutações no gene TCOF1 (no locus cromossômico 5q31.3-33.3), que codifica uma fosfoproteína nucleolar responsável pela formação da parte craniofacial do embrião humano. Como resultado de uma diminuição prematura na quantidade dessa proteína, a biogênese e as funções do rRNA são interrompidas. De acordo com geneticistas do programa de pesquisa Genoma Humano, esses processos levam à redução da proliferação de células embrionárias da crista neural – uma crista ao longo do sulco neural, que se fecha em um tubo neural durante o desenvolvimento embrionário.
A formação dos tecidos faciais ocorre devido à transformação e diferenciação de células da parte superior (cabeça) da crista neural, que migram ao longo do tubo neural para a área do primeiro e segundo arcos branquiais do embrião. A deficiência dessas células causa deformações craniofaciais. O período crítico para a ocorrência de anomalias é de 18 a 28 dias após a fertilização. Após a conclusão da migração das células da crista neural (na quarta semana de gestação), quase todos os tecidos mesenquimais frouxos da região facial são formados, os quais posteriormente (de 5 a 8 semanas) se diferenciam em tecidos esqueléticos e conjuntivos de todas as partes da face, pescoço, laringe, orelha (incluindo a orelha interna) e futuros dentes.
Patogênese
A patogênese da síndrome de Treacher Collins é frequentemente familiar, e a anomalia é herdada de forma autossômica dominante, embora existam casos de transmissão autossômica recessiva do defeito (com mutações em outros genes, em particular POLR1C e POLR1D). O aspecto mais imprevisível da disostose maxilofacial é que a mutação é herdada por crianças em apenas 40-48% dos casos. Ou seja, em 52-60% dos pacientes, as causas da síndrome de Treacher Collins não estão associadas à presença de uma anomalia na família, e acredita-se que a patologia ocorra como resultado de mutações genéticas esporádicas de novo. Muito provavelmente, novas mutações são consequências de efeitos teratogênicos no feto durante a gravidez.
Entre as causas teratogênicas dessa síndrome, os especialistas citam altas doses de etanol (álcool etílico), radiação, fumaça de cigarro, citomegavírus e toxoplasmose, além de herbicidas à base de glifosato (Roundal, Glyfor, Tornado, etc.). A lista de fatores iatrogênicos inclui medicamentos para acne e seborreia com ácido 13-cis-retinóico (isotretinoína, Accutane); o anticonvulsivante fenitoína (Dilantin, Epanutin); e os psicotrópicos diazepam, Valium, Relanium, Seduxen.
Sintomas Síndrome de Treacher Collins
Em sua maioria, os sinais clínicos da disostose mandibulofascial e o grau de sua expressão dependem das características da manifestação das mutações genéticas. E os primeiros sinais dessa anomalia, na maioria dos casos, são visíveis na criança imediatamente após o nascimento: o rosto com a síndrome de Treacher Collins tem uma aparência característica. Além disso, as anomalias morfológicas são geralmente bilaterais e simétricas.
Os sintomas mais óbvios da síndrome de Treacher Collins são:
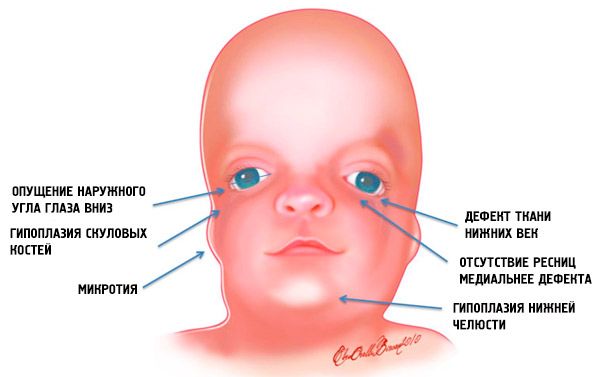
- subdesenvolvimento (hipoplasia) dos ossos faciais do crânio: zigomático, processos zigomáticos do osso frontal, placas pterigóides laterais, seios paranasais, maxilar inferior e saliências das epífises ósseas (côndilos);
- subdesenvolvimento dos ossos do maxilar inferior (micrognatia) e um ângulo mandibular mais obtuso do que o normal;
- o nariz é de tamanho normal, mas parece grande devido à hipoplasia dos arcos superciliares e ao subdesenvolvimento ou ausência dos arcos zigomáticos na região temporal;
- as fendas dos olhos são para baixo, ou seja, o formato dos olhos é anormal, com os cantos externos caídos para baixo;
- defeitos das pálpebras inferiores (coloboma) e ausência parcial de cílios;
- aurículas de formato irregular com uma ampla gama de desvios, incluindo sua localização no canto da mandíbula inferior, ausência de lóbulos, fístulas cegas entre o trago da orelha e o canto da boca, etc.;
- estreitamento ou fechamento (atresia) do canal auditivo externo e anomalias dos ossículos do ouvido médio;
- ausência ou hipoplasia das glândulas salivares parótidas;
- hipoplasia faríngea (estreitamento da faringe e das vias aéreas);
- não fusão do palato duro (fenda palatina), bem como ausência, encurtamento ou imobilidade do palato mole.
Tais anomalias anatômicas, em todos os casos, apresentam complicações. Trata-se de distúrbios auditivos funcionais, como perda auditiva condutiva ou surdez completa; deficiência visual devido à formação inadequada dos globos oculares; defeitos do palato que causam dificuldades na alimentação e na deglutição. Existem distúrbios de oclusão dentária (má oclusão) associados a defeitos mandibulares, que, por sua vez, causam problemas na mastigação e na articulação. Patologias do palato mole explicam a voz anasalada.
Complicações e consequências
As consequências das anomalias maxilofaciais na síndrome de Treacher Collins são que, ao nascer, as capacidades intelectuais da criança são normais, mas devido a defeitos auditivos e outros distúrbios, observa-se retardo mental secundário.
Além disso, crianças com tais defeitos sentem intensamente sua inferioridade e sofrem, o que afeta negativamente seu sistema nervoso e sua psique.
Diagnósticos Síndrome de Treacher Collins
O diagnóstico pós-natal da síndrome de Treacher Collins baseia-se essencialmente nos sinais clínicos. A disostose craniofacial é facilmente identificada quando a síndrome é totalmente expressiva, mas quando há sintomas patológicos minimamente expressos, podem surgir dificuldades para estabelecer o diagnóstico correto.
Neste caso, atenção especial deve ser dada à avaliação de todas as funções associadas às anomalias, especialmente aquelas que afetam a respiração (devido ao risco de apneia do sono). A eficácia da alimentação e a saturação de oxigênio da hemoglobina também devem ser avaliadas e monitoradas.
Mais tarde, no 5º ou 6º dia após o nascimento, a extensão do dano auditivo precisará ser determinada por meio de testes audiológicos, que devem ser realizados na maternidade.
É prescrito um exame, durante o qual são realizados diagnósticos instrumentais por fluoroscopia da dismorfologia craniofacial; pantomografia (raio-X panorâmico das estruturas ósseas do crânio facial); tomografia computadorizada craniana completa em várias projeções; tomografia computadorizada ou ressonância magnética do cérebro para determinar o estado do canal auditivo interno.
O diagnóstico mais precoce – pré-natal – de anomalias maxilofaciais na presença da síndrome de Treacher Collins na história familiar é possível por meio de biópsia de vilo corial entre 10 e 11 semanas de gestação (o procedimento pode causar aborto espontâneo e infecção do útero).
Exames de sangue também são feitos com familiares; entre 16 e 17 semanas de gestação, o líquido amniótico é analisado (amniocentese transabdominal); entre 18 e 20 semanas de gestação, é realizada a fetoscopia e o sangue é coletado dos vasos fetais da placenta.
Mas, na maioria das vezes, o ultrassom é usado no diagnóstico pré-natal dessa síndrome no feto (entre 20 e 24 semanas de gestação).
Quais testes são necessários?
Diagnóstico diferencial
Esses mesmos métodos são usados por especialistas quando diagnósticos diferenciais são necessários para reconhecer a síndrome de Treacher Collins leve e distingui-la de outras anomalias congênitas dos ossos craniofaciais, em particular: síndromes de Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph, bem como microsomia hemifacial (síndrome de Goldenhar), hipertelorismo, fusão prematura das suturas cranianas (craniossinostose) ou fusão prejudicada dos ossos faciais (craniossinostose).
Quem contactar?
Tratamento Síndrome de Treacher Collins
Como em todos os casos de defeitos congênitos geneticamente determinados, o tratamento das formas graves da síndrome de Treacher Collins é exclusivamente paliativo, visto que simplesmente não existem métodos terapêuticos para tais patologias. O espectro e o grau de deformidades nessa síndrome são extensos e, portanto, a natureza e a intensidade da intervenção médica também oferecem muitas opções.
Aparelhos auditivos são usados para corrigir e melhorar a audição, e sessões de terapia da fala são usadas para melhorar a fala.
Intervenções cirúrgicas são necessárias em idade precoce em casos graves de estreitamento das vias aéreas (traqueostomia) e da laringe (gastrostomia para alimentação). A correção cirúrgica do palato também pode ser necessária.
As cirurgias de alongamento mandibular são realizadas a partir dos 2 a 3 anos de idade. A reconstrução de tecidos moles inclui correção de coloboma da pálpebra inferior e cirurgia plástica auricular.
Prevenção
A prevenção da síndrome de Treacher Collins envolve o comparecimento dos futuros pais a aconselhamento genético e, se houver histórico familiar da síndrome, surge a questão da possibilidade de gravidez em si – para evitar o nascimento de uma criança com anomalias craniofaciais.
Previsão
Qual é o prognóstico para esta patologia? Depende do grau de deformação e da intensidade dos sintomas. A síndrome de Treacher Collins é um diagnóstico para toda a vida.
 [ 25 ]
[ 25 ]