Médico especialista do artigo

Novas publicações
Síndrome de Martin-Bell
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
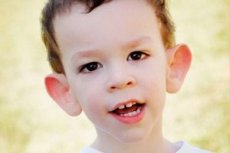
A síndrome de Martin-Bell foi descrita em 1943 por médicos, que lhe deram o nome. A doença é um distúrbio genético que consiste em retardo mental. Em 1969, foram identificadas alterações no cromossomo X (fragilidade na extremidade distal do braço), características dessa doença. Em 1991, cientistas descobriram o gene responsável pelo desenvolvimento da doença. Essa doença também é chamada de "síndrome do X frágil". Tanto meninos quanto meninas são suscetíveis à doença, mas os meninos são mais frequentemente afetados (3 vezes).
Epidemiologia
A síndrome de Martin-Bell é uma doença bastante comum: 0,3 a 1 em cada 1.000 homens sofrem desta doença, e 0,2 a 0,6 em cada 1.000 mulheres. Além disso, crianças com síndrome de Martin-Bell nascem em todos os continentes com a mesma frequência. Obviamente, nacionalidade, cor da pele, formato dos olhos, condições de vida e bem-estar das pessoas não afetam a ocorrência da doença. Sua frequência de ocorrência é comparável apenas à frequência da síndrome de Down (1 em cada 600 a 800 recém-nascidos). Um quinto dos portadores do gene alterado são saudáveis, não apresentam anormalidades clínicas ou genéticas, e o restante apresenta sinais de retardo mental, de formas leves a graves. Entre as portadoras do gene alterado, pouco mais de um terço está doente.
A síndrome do X frágil afeta aproximadamente 1 em cada 2.500 a 4.000 homens e 1 em cada 7.000 a 8.000 mulheres. A prevalência de portadores entre mulheres é estimada em 1 em 130 a 250; a prevalência de portadores entre homens é estimada em 1 em 250 a 800.
Causas Síndrome de Martin-Bell
A síndrome de Martin-Bell se desenvolve devido à cessação completa ou parcial da produção de uma proteína específica pelo corpo. Isso ocorre devido à falta de resposta do gene FMR1, localizado no cromossomo X. A mutação ocorre como resultado da reestruturação do gene a partir de variantes estruturais instáveis dos estados gênicos (alelos), e não desde o início. A doença é transmitida apenas pela linhagem masculina, e o homem pode não estar necessariamente doente. Os portadores do sexo masculino transmitem o gene para suas filhas de forma inalterada, portanto, seu retardo mental não é óbvio. Com a transmissão posterior do gene da mãe para os filhos, o gene sofre mutação, e todos os sinais característicos da doença aparecem.
Patogênese
A patogênese da síndrome de Martin-Bell baseia-se em mutações no aparato genético, que levam ao bloqueio da produção da proteína FMR, uma proteína vital para o corpo, especialmente nos neurônios, e presente em vários tecidos. Pesquisas mostram que as proteínas FMR estão diretamente envolvidas nos processos de regulação das traduções que ocorrem no tecido cerebral. A ausência dessa proteína ou sua produção limitada pelo corpo leva ao retardo mental.
Na patogênese da doença, a hipermetilação gênica é considerada um distúrbio chave, mas ainda não foi possível identificar definitivamente o mecanismo de desenvolvimento desse distúrbio.
Ao mesmo tempo, também foi descoberta a heterogeneidade do locus da patologia, associada ao polialelismo, bem como ao polilocus. Foi determinada a presença de variantes alélicas no desenvolvimento da doença, causadas pela existência de mutações pontuais, bem como pela destruição do gene do tipo FMRL.
Os pacientes também apresentam dois tripletos frágeis sensíveis ao ácido fólico, localizados a 300 kb, bem como a 1,5-2 milhões de pares de bases do tripleto frágil que contém o gene FMR1. O mecanismo das mutações que ocorrem nos genes FRAXE e FRAXF (identificados nos tripletos frágeis mencionados) está relacionado ao mecanismo dos distúrbios da síndrome de Martin Bell. Esse mecanismo é causado pela disseminação das repetições GCC e CGG, que causam a metilação das chamadas ilhas CpG. Além da forma clássica da patologia, existem também dois tipos raros que se diferenciam pela expansão das repetições trinucleotídicas (na meiose masculina e feminina).
Constatou-se que, na forma clássica da síndrome, o paciente não possui uma proteína nucleocitoplasmática específica do tipo FMR1, que desempenha a função de se ligar a vários mRNAs. Além disso, essa proteína promove a formação de um complexo que auxilia nos processos de tradução dentro dos ribossomos.
Sintomas Síndrome de Martin-Bell
Como reconhecer a doença em crianças? Quais são os primeiros sinais? Nos primeiros meses de vida de uma criança, é impossível reconhecer o sintoma de Martin-Bell, exceto que às vezes se observa uma diminuição do tônus muscular. Após um ano, o quadro clínico da doença torna-se mais evidente: a criança começa a andar e a falar tardiamente, às vezes a fala está completamente ausente. Ela é hiperativa, agita os braços aleatoriamente, tem medo de multidões e barulho, é teimosa, tem explosões agudas de raiva, instabilidade emocional, ocorrem crises epilépticas e não faz contato visual. Em pacientes com síndrome de Martin-Bell, a doença também se manifesta pela aparência: orelhas proeminentes e grandes, testa pesada, rosto alongado, queixo proeminente, estrabismo, mãos e pés largos. Distúrbios endócrinos também são característicos: frequentemente grande peso, obesidade, testículos grandes em homens, puberdade precoce.

Entre os pacientes com a síndrome de Martin-Bell, o nível de inteligência varia muito: desde retardo mental leve até casos graves. Se uma pessoa normal tem um quociente de inteligência (QI) de 100 em média, e um gênio tem 130, então pessoas suscetíveis à doença têm de 35 a 70.
Todos os sintomas clínicos da patologia podem ser caracterizados por uma tríade de manifestações principais:
- oligofrenia (QI é 35-50);
- dismorfofobia (observam-se orelhas em abano e prognatismo);
- macroorquidismo, que aparece após o início da puberdade.
Aproximadamente 80% dos pacientes também apresentam prolapso da válvula bicúspide.
No entanto, a forma completa da síndrome se manifesta em apenas 60% dos pacientes. Em 10%, apenas o retardo mental é detectado e, no restante, a doença se desenvolve com uma combinação diferente de sintomas.
Entre os primeiros sinais da doença, que aparecem em idade precoce:
- a criança doente apresenta retardo mental significativo em comparação ao desenvolvimento de outros pares;
- distúrbios de atenção e concentração;
- forte teimosia;
- as crianças começam a andar e a falar bastante tarde;
- são observados distúrbios de hiperatividade e desenvolvimento da fala;
- crises de raiva muito fortes e incontroláveis;
- pode ocorrer mutismo - uma ausência completa da fala em uma criança;
- o bebê sente ansiedade social e é capaz de entrar em pânico devido a barulhos altos ou quaisquer outros sons altos;
- a criança agita os braços de forma incontrolável e caótica;
- observa-se timidez, a criança tem medo de ficar em locais lotados;
- o surgimento de várias ideias obsessivas, estado emocional instável;
- O bebê pode relutar em fazer contato visual com as pessoas.
Em adultos, são observados os seguintes sintomas da patologia:
- aparência específica: rosto alongado com testa pesada, orelhas grandes e proeminentes, queixo fortemente proeminente;
- pés chatos, otites e estrabismo;
- a puberdade ocorre bem cedo;
- pode desenvolver obesidade;
- Muitas vezes, defeitos cardíacos são observados na síndrome de Martin-Bell;
- nos homens, observa-se aumento dos testículos;
- as articulações das articulações tornam-se muito móveis;
- o peso e a altura aumentam acentuadamente.
Diagnósticos Síndrome de Martin-Bell
Para diagnosticar a síndrome de Martin Bell, você precisa consultar um geneticista qualificado. O diagnóstico é feito após testes genéticos específicos que permitem identificar o cromossomo defeituoso.
Testes
Em um estágio inicial da doença, utiliza-se um método citogenético, no qual um fragmento de material celular é retirado do paciente, ao qual se adiciona ácido fólico para provocar alterações nos cromossomos. Após um certo período, identifica-se uma área do cromossomo onde há um afinamento perceptível – este é um sinal da presença da síndrome do cromossomo X frágil.
Entretanto, esse teste não é adequado para diagnóstico em estágios mais avançados da doença, porque sua precisão é reduzida pelo uso generalizado de multivitamínicos contendo ácido fólico.
O diagnóstico integrado da síndrome de Martin-Bell é um exame genético molecular, que consiste na determinação do número de repetições chamadas trinucleotídeos no gene.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnóstico instrumental
Um método altamente específico de diagnóstico instrumental é a PCR (reação em cadeia da polimerase), que permite estudar a estrutura dos resíduos de aminoácidos contidos no cromossomo X e, assim, determinar a presença da síndrome de Martin Bell.
Existe também um método separado, ainda mais específico, de diagnóstico de patologias: uma combinação de PCR e detecção por eletroforese capilar. Este método é altamente preciso e detecta patologias cromossômicas em pacientes com insuficiência ovariana primária, bem como síndrome atáxica.
A presença do defeito pode ser determinada após a realização de diagnóstico por EEG. Pacientes com esta doença apresentam atividade cerebral bioelétrica semelhante.
Quais testes são necessários?
Diagnóstico diferencial
Métodos diferenciados que ajudam a suspeitar da síndrome incluem:
- clínico - 97,5% dos pacientes apresentam sinais óbvios de retardo mental (moderado ou profundo); 62% têm orelhas grandes e proeminentes; 68,4% têm queixo e testa grandes e proeminentes; 68,4% dos meninos têm testículos aumentados, 41,4% têm peculiaridades na fala (velocidade de fala irregular, volume incontrolável, etc.);
- citogênico - o sangue é examinado para cultura de linfócitos, é determinado o número de células com cromossomo X frágil por 100 células estudadas;
- Eletroencefalografia - são registradas alterações nos impulsos elétricos do cérebro específicas da síndrome de Martin-Bell.
Quem contactar?
Tratamento Síndrome de Martin-Bell
No tratamento de pacientes adultos, são utilizados antidepressivos com psicoestimulantes. O processo de terapia medicamentosa é constantemente monitorado por psicólogo e psiquiatra. Além disso, procedimentos de microinjeção com medicamentos como Cerebrolysin (ou seus derivados), bem como citomedinas (como Solcoseryl ou Lidase), são realizados em clínicas particulares.
No desenvolvimento da síndrome atáxica, são utilizados medicamentos anticoagulantes e nootrópicos. Além disso, são prescritas misturas de aminoácidos e angioprotetores. Mulheres com insuficiência ovariana primária recebem tratamento corretivo com fitoterápicos e estrogênios.
Antagonistas do receptor de glutamina também são usados no tratamento.
Tradicionalmente, o tratamento da síndrome de Martin-Bell envolve o uso de medicamentos que afetam os sintomas da doença, mas não sua causa. Essa terapia envolve a prescrição de antidepressivos, neurolépticos e psicoestimulantes. Nem todos os medicamentos são indicados para uso em crianças, portanto, a lista de medicamentos é bastante limitada. Os neurolépticos que podem ser usados após os 3 anos (a idade mínima para prescrição) incluem haloperidol em gotas e comprimidos, clorpromazina em solução e periciazina em gotas. Assim, a dose de haloperidol para crianças é calculada dependendo do peso corporal. Para adultos, a dose é prescrita individualmente. É administrado por via oral, começando com 0,5 a 5 mg, 2 a 3 vezes ao dia, e então a dose é gradualmente aumentada para 10 a 15 mg. Quando ocorre melhora, eles mudam para uma dose mais baixa para manter a condição alcançada. Em caso de agitação psicomotora, 5 a 10 mg são prescritos por via intramuscular ou intravenosa, várias repetições são possíveis após 30 a 40 minutos. A dose diária não deve exceder 100 mg. Podem ocorrer efeitos colaterais como náuseas, vômitos, espasmos musculares, aumento da pressão, arritmia, etc. Idosos devem tomar precauções especiais, pois já foram registrados casos de parada cardíaca súbita e pode ocorrer discinesia tardia (movimentos involuntários).
Os antidepressivos aumentam a atividade das estruturas cerebrais, aliviam a depressão, a tensão e melhoram o humor. Esses medicamentos, recomendados para uso a partir dos 5-8 anos de idade para a síndrome de Martin-Bell, incluem clomipromina, sertralina, fluoxetina e fluvoxamina. Portanto, a fluoxetina é administrada por via oral durante as refeições, 1-2 vezes ao dia (de preferência na primeira metade do dia), começando com 20 mg por dia, aumentando para 80 mg, se necessário. Doses superiores a 60 mg não são recomendadas para idosos. O tratamento é determinado pelo médico, mas não deve exceder 5 semanas.
Possíveis efeitos colaterais: tontura, ansiedade, zumbido, perda de apetite, taquicardia, edema, etc. É necessária cautela ao prescrever para idosos, pessoas com doenças cardiovasculares e diabetes.
Psicoestimulantes são drogas psicotrópicas usadas para melhorar a percepção de estímulos externos: eles aguçam a audição, as reações de resposta e a visão.
O diazepam é prescrito como sedativo para neuroses, ansiedade, crises epilépticas e convulsões. É administrado por via oral, intravenosa, intramuscular e retal (no reto). É prescrito individualmente, dependendo da gravidade da doença, com doses mínimas de 5 a 10 mg e doses diárias de 5 a 20 mg. A duração do tratamento é de 2 a 3 meses. Para crianças, a dose é calculada levando em consideração o peso corporal e as características individuais. Os efeitos colaterais incluem letargia, apatia, sonolência, náusea e constipação. É perigoso combiná-lo com álcool, pois pode causar dependência.
No tratamento da síndrome de Martin-Bell, houve casos de melhora da condição com a introdução de medicamentos à base de material animal (cérebro): cerebrolisato, cerebrolisina, cerebrolisato-M. Os principais componentes desses medicamentos são peptídeos que promovem a produção de proteínas nos neurônios, repondo assim a proteína em falta. A cerebrolisina é administrada em um jato de 5 a 10 ml, e o tratamento consiste em 20 a 30 injeções. O medicamento é prescrito para crianças a partir de um ano de idade, administrado por via intramuscular, diariamente, na dose de 1 a 2 ml, durante um mês. Sessões repetidas de administração são possíveis. Os efeitos colaterais incluem febre e são contraindicados para gestantes.
Houve tentativas de tratar a doença com ácido fólico, mas apenas o aspecto comportamental melhorou (o nível de agressividade e hiperatividade diminuiu, a fala melhorou) e nada mudou no nível intelectual. Para melhorar o quadro da doença, prescreve-se ácido fólico, e são indicados métodos de fisioterapia, fonoaudiologia e correção pedagógica e social.
Preparações de lítio também são consideradas eficazes, pois ajudam a melhorar a adaptação do paciente ao ambiente social, bem como a atividade cognitiva. Além disso, regulam seu comportamento em sociedade.
O uso de ervas para a síndrome de Martin-Bell é possível como antidepressivos. Ervas que ajudam a aliviar a tensão, a ansiedade e a melhorar o sono incluem valeriana, hortelã-pimenta, tomilho, erva-de-são-joão e camomila. As infusões são preparadas da seguinte forma: para 1 colher de chá de ervas secas, você precisará de um copo de água fervente. As decocções são infundidas por pelo menos 20 minutos, tomadas principalmente à noite, antes de dormir, ou à tarde. Uma colher de mel seria um bom complemento.
Tratamento de fisioterapia
Para eliminar as manifestações neurológicas, são realizados procedimentos fisioterapêuticos especiais - como exercícios em piscina, relaxamento muscular e acupuntura.
Tratamento cirúrgico
Uma etapa importante do tratamento também é considerada a cirurgia plástica – operações que ajudam a melhorar a aparência do paciente. Realiza-se cirurgia plástica dos membros e das orelhas, bem como dos genitais. Também é realizada a correção de ginecomastia com epispádia, bem como de outros defeitos na aparência.
Prevenção
O único método de prevenção da doença é o rastreamento pré-natal de gestantes. Existem exames específicos que permitem a detecção precoce da patologia, após os quais se recomenda a interrupção da gravidez. Como alternativa, utiliza-se a fertilização in vitro, que pode ajudar a criança a herdar um cromossomo X saudável.
A prevenção do paciente depende se a mutação genética surgiu novamente ou foi herdada. Para isso, são realizados diagnósticos genéticos moleculares. O fato de o teste não ter revelado o "cromossomo X frágil" em parentes fala a favor da "frescura" da mutação, o que significa que o risco de ter um filho com a síndrome de Martin-Bell é muito pequeno. Em famílias com pessoas doentes, o teste ajudará a evitar a recorrência de casos.
Previsão
O prognóstico da síndrome de Martin-Bell é favorável para a vida toda, mas não para a recuperação. A expectativa de vida depende da gravidade da doença e dos defeitos associados. O paciente pode viver uma vida normal. Nas formas graves da síndrome de Martin-Bell, os pacientes correm o risco de incapacidade permanente.
Expectativa de vida
A síndrome de Martin Bell não tem um impacto negativo sério na saúde, portanto, a expectativa de vida da maioria das pessoas diagnosticadas com essa patologia não difere dos indicadores padrão.