Médico especialista do artigo

Novas publicações
Polimiosite e dermatomiosite: causas, sintomas, diagnóstico, tratamento
Última revisão: 23.04.2024

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
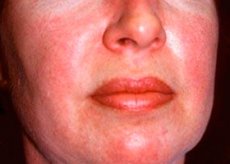
A polimiosite e a dermatomiosite são doenças reumáticas sistêmicas raras , caracterizadas por alterações musculares inflamatórias e degenerativas (polimiosite) ou músculos e pele (dermatomiosite). A manifestação cutânea mais específica é uma erupção heliotrópica.
As lesões musculares são simétricas e incluem fraqueza, alguma dor e posterior atrofia dos músculos proximais do cinturão da extremidade superior. As complicações podem incluir danos nos órgãos internos e malignidades. O diagnóstico baseia-se na análise do quadro clínico e na avaliação dos distúrbios musculares, determinando as concentrações das enzimas relevantes, realizando RM, eletromiografia e biópsia do tecido muscular. O tratamento utiliza glicocorticóides, às vezes em combinação com imunossupressores ou imunoglobulinas administradas por via intravenosa.
As mulheres estão doentes duas vezes mais que os homens. A doença pode ocorrer em qualquer idade, mas é mais frequentemente detectada no intervalo de 40 a 60 anos; em crianças de 5 a 15 anos.
 [
[O que causa dermatomiosite e polimiosite?
A causa da doença é suposto ser uma reação auto-imune ao tecido muscular em indivíduos geneticamente predispostos. A doença é mais comum na presença de uma história familiar sobrecarregada e portadores de alguns antígenos HLA (DR3, DR52, DR56). Possíveis fatores de inicialização são miosite viral e neoplasias malignas. Há relatos da detecção em células musculares de estruturas semelhantes aos picornavírus; Além disso, os vírus podem induzir doenças semelhantes em animais. A associação de tumores malignos com dermatomiosite (muito menos do que com polimiosite) sugere que o crescimento tumoral também pode ser um mecanismo de gatilho para o desenvolvimento da doença como resultado de desencadear reações auto-imunes a antígenos tumorais e musculares comuns.
Nas paredes dos vasos sanguíneos dos músculos esqueléticos, são detectados depósitos de IgM, IgG e o terceiro componente do complemento; Isto é especialmente verdadeiro em crianças com dermatomiosite. Pacientes com polimiosite também podem desenvolver outros processos auto-imunes.
Dermatomiosite e polimiosite de fisiopatologia
As alterações patológicas incluem danos celulares e sua atrofia contra um fundo de inflamação de severidade variável. Os músculos das extremidades superiores e inferiores, bem como o rosto, são menos afetados do que outros músculos esqueléticos. A derrota da musculatura visceral da faringe e das partes superiores do esôfago, menos freqüentemente do coração, do estômago ou do intestino, pode levar a uma ruptura nas funções desses órgãos. Altas concentrações de mioglobina, causadas por rabdomiólise, podem causar danos nos rins. Também pode haver alterações inflamatórias nas articulações e nos pulmões, especialmente em pacientes que possuem anticorpos anti-sintetizados.
Sintomas de dermatomiosite e polimiosite
O início da polimiosite pode ser agudo (especialmente em crianças) ou subaguda (geralmente em adultos). A infecção viral aguda às vezes precede ou é o fator inicial da manifestação da doença, cujas manifestações mais freqüentes são a fraqueza dos músculos proximais ou erupções cutâneas. As sensações de dor são menos expressas do que a fraqueza. Talvez o desenvolvimento da poliartralgia, o fenômeno de Raynaud, a disfagia, as violações dos pulmões, sintomas comuns (febre, redução da massa, fraqueza). O fenômeno de Reynaud é freqüentemente encontrado em pacientes com doenças concomitantes do tecido conjuntivo.
A fraqueza muscular pode progredir por várias semanas ou meses. No entanto, para a manifestação clínica da fraqueza muscular, um mínimo de 50% das fibras musculares deve ser afetada (assim, a presença de fraqueza muscular indica progressão da miosite). Os pacientes podem ter dificuldade em levantar os braços acima do nível do ombro, subindo as escadas, saindo da sessão. Devido à fraqueza pronunciada dos músculos pélvicos e da cintura escapular, os pacientes podem ser rebitados em uma cadeira de rodas ou cama. Com a derrota do flexor do pescoço, torna-se impossível arrancar a cabeça do travesseiro. A derrota dos músculos da faringe e das partes superiores do esôfago leva a uma violação da deglutição e regurgitação. Os músculos dos membros inferiores, superiores e do rosto geralmente não são afetados. No entanto, o desenvolvimento de contraturas de membros é possível.
As erupções cutâneas, observadas com dermatomiosite, geralmente apresentam cor escura e caráter eritematoso. O inchaço periorbital de cor roxa (erupção heliotrópica) também é característico. As erupções cutâneas podem aumentar ligeiramente acima do nível da pele e ser lisas ou cobertas com escamas; localização de lesões - a testa, pescoço, ombros, peito, costas, antebraços, partes inferiores das sobrancelhas coxas, na área do joelho, o maléolo medial, a superfície traseira do interfalângica e articulações metacarpofalângicas, com a face lateral (Gottrona de sintomas). Possível hiperemia da base ou periferia das unhas. Na pele da superfície lateral dos dedos, é possível desenvolver dermatite desquamativa, acompanhada pelo aparecimento de rachaduras. As lesões cutâneas primárias são mais frequentemente resolvidas sem consequências, mas podem levar ao desenvolvimento de alterações secundárias sob a forma de pigmentação escura, atrofia, cicatrizes ou vitiligo. Possível formação de calcificações subcutâneas, especialmente em crianças.
Aproximadamente 30% dos pacientes desenvolvem poliartralgia ou poliartrite, muitas vezes acompanhada de inchaço e derrame articular. No entanto, a gravidade das manifestações articulares é baixa. Mais frequentemente, eles ocorrem quando os pacientes possuem anticorpos para Jo-1 ou outras sintetases.
A derrota dos órgãos internos (com exceção da faringe e esôfago superior) em polimiosite é menos comum do que em outras doenças reumáticas (em particular, SLE e esclerodermia sistêmica). Raramente, especialmente com uma síndrome antisintética, a doença se manifesta como uma pneumonia intersticial (na forma de dispneia e tosse). Podem desenvolver-se arritmias cardíacas e distúrbios de condução, mas geralmente são assintomáticos. Manifestações do trato gastrointestinal são mais comuns em crianças que também sofrem de vasculite e podem incluir vômitos com uma mistura de sangue, melena e perfuração do intestino.
Aonde dói?
Classificação da polimiosite
Existem 5 variantes de polimiosite.
- Polimiosite idiopática primária, que pode ocorrer em qualquer idade. Com isso, não há lesão na pele.
- A dermatomiosite idiopática primária é semelhante à polimiosite primária idiopática, mas com ela há uma lesão da pele.
- A polimiosite e a dermatomiosite associada a neoplasias malignas podem ocorrer em pacientes de qualquer idade; mais freqüentemente observado em pacientes idosos, bem como em pacientes com outras doenças do tecido conjuntivo. O desenvolvimento de neoplasias malignas pode ser observado dentro de 2 anos antes e dentro de 2 anos após a estréia da miosite.
- A polimiosite infantil ou dermatomiosite está associada a vasculite sistêmica.
- A polimiosite e a dermatomiosite também podem ocorrer em pacientes que sofrem de outras doenças do tecido conjuntivo, na maioria das vezes com esclerose sistêmica progressiva, doença mista do tecido conjuntivo e LES.
A inclusão do músculo do tronco no grupo da polimiosite da miosite é incorreta, uma vez que esta é uma doença separada caracterizada por manifestações clínicas semelhantes às da polimiosite crônica idiopática. No entanto, ele se desenvolve em idade avançada, afeta geralmente os músculos das partes distal do corpo (por exemplo, extremidades superiores e inferiores), tem uma duração maior, responde mal ao tratamento e é caracterizada por uma imagem histológica típica.
Diagnóstico de dermatomiosite e polimiosite
Deve-se suspeitar de poliomiosite em pacientes com queixa de fraqueza dos músculos proximais, acompanhados de sua dor ou sem ele. O exame de dermatomiosite é necessário para pacientes queixam-se de erupções parecidas com sintoma de heliotrópio ou Gotstrom, bem como em pacientes com manifestações de polimiosite, combinadas com lesões cutâneas que correspondem a dermatomiosite. As manifestações clínicas de polimiosite e dermatomiosite podem assemelhar-se àqueles de esclerose sistêmica ou, menos freqüentemente, LES ou vasculite. A confiabilidade do diagnóstico é aumentada ao combinar o maior número possível desses cinco critérios:
- fraqueza dos músculos proximais;
- erupções cutâneas características;
- aumento da atividade das enzimas dos tecidos musculares (creatina quinase ou, na ausência de aumento na atividade, aminotransferases ou aldolase);
- alterações características na miografia ou na ressonância magnética;
- alterações histológicas características na biópsia do tecido muscular (critério absoluto).
Uma biópsia muscular pode excluir algumas condições clinicamente semelhantes, como músculo do tronco muscular e rabdomiólise causada por uma infecção viral. As alterações reveladas pelo exame histológico podem ser diferentes, mas típicas são inflamação crônica, focos de degeneração e regeneração dos músculos. Antes do início do tratamento potencialmente tóxico, um diagnóstico preciso deve ser feito (geralmente por verificação histológica). Com a ressonância magnética, é possível identificar focos de edema e inflamação nos músculos seguidos por uma biópsia alvo.
Os testes laboratoriais permitem reforçar ou, pelo contrário, eliminar a suspeita da presença da doença e também útil na avaliação da gravidade, a possibilidade de se combinar com outra patologia similar e diagnosticar complicações. Apesar de os anticorpos antinucleares serem detectados em alguns pacientes, esse fenômeno é mais típico para outras doenças do tecido conjuntivo. Cerca de 60% dos pacientes possuem anticorpos contra o antígeno nuclear (PM-1) ou células inteiras do timo e Jo-1. O papel dos autoanticorpos na patogênese da doença ainda não é claro, embora se saiba que os anticorpos para Jo-1 são um marcador específico de uma síndrome antisintética, incluindo alveolite fibrosante, fibrose pulmonar, artrite e fenômeno de Reino.
A avaliação periódica da atividade da creatina quinase é útil para o monitoramento do tratamento. No entanto, com atrofia muscular severa, a atividade da enzima pode ser normal, apesar da presença de miosite crônica ativa. Os dados de ressonância magnética, as biópsias musculares ou os valores elevados de atividade da creatina quinase ajudam a diferenciar a recorrência da polimiosite e miopatia induzida por glicocorticóides.
Uma vez que muitos pacientes têm neoplasmas malignos não diagnosticados, alguns autores recomendam o rastreio de todos os adultos com dermatomiosite e pessoas com polimiosite com idade superior a 60 anos de acordo com o seguinte esquema: exame físico, incluindo exame mamário, exame ginecológico e exame do reto ( incluindo o estudo de fezes para o sangue latente); teste de sangue clínico; exames de sangue bioquímicos; mamografia; determinação do antígeno embrionário do câncer; análise geral da urina; radiografia de tórax. A necessidade de tal rastreio para pacientes mais jovens que não possuem sinais clínicos de tumores malignos foi questionada por alguns autores.
O que precisa examinar?
Tratamento de dermatomiosite e polimiosite
Antes de interromper a inflamação, é necessário limitar a atividade física. Os glucocorticóides são drogas de primeira linha. Na fase aguda da doença, pacientes adultos precisam de prednisolona (dentro) em uma dose de 40 a 60 mg por dia. A detecção regular da atividade da creatina quinase é um indicador precoce de eficácia: na maioria dos pacientes, sua diminuição ou normalização ocorre dentro de 6 a 12 semanas após o aumento da força muscular. Após a normalização da atividade enzimática, a dose de prednisolona diminui: primeiro cerca de 2,5 mg por dia durante a semana, em seguida, mais rapidamente; Quando a atividade das enzimas musculares aumenta, a dose do hormônio é novamente aumentada. Os pacientes recuperados podem fazer sem glicocorticóides, mas, mais frequentemente, os pacientes adultos precisam de terapia com glicocorticóide a longo prazo (10-15 mg de prednisolona por dia). A dose inicial de prednisolona para crianças é de 30-60 mg / m 2, uma vez por dia. Se houver uma remissão> 1 ano, as crianças podem ter uma cessação da terapia com glicocorticóides.
Em alguns casos, em pacientes que recebem altas doses de glicocorticóides, ocorre aumento repentino da fraqueza muscular, o que pode ser devido ao desenvolvimento de miopatia com glicocorticóide.
Quando resposta inadequada a um tratamento glucocorticóide e também no desenvolvimento de miopatia glucocorticóide ou outras complicações que requerem uma redução da dose ou retirada prednisolona, utilizar imunosupressores (metotrexato, ciclofosfamida, azatioprina, ciclosporina). Alguns pacientes podem receber apenas metotrexato (geralmente em doses superiores aos do tratamento da AR) por mais de 5 anos. As imunoglobulinas intravenosas podem ser eficazes em pacientes refratários à terapia medicamentosa, mas seu uso aumenta o custo do tratamento.
A miosite associada a tumores primários e metastáticos, bem como a miosite dos músculos do tronco, geralmente são mais refratárias à terapia com glucocorticoides. O desenvolvimento da remissão associada a tumores malignos de miosite é possível após a remoção do tumor.
Qual o prognóstico da dermatomiosite e da polimiosite?
A remissão a longo prazo (e até a recuperação clínica) durante 5 anos é observada em mais da metade dos pacientes tratados; Em crianças, este indicador é maior. A recaída, no entanto, pode se desenvolver a qualquer momento. A taxa global de sobrevivência de cinco anos é de 75%, maior em crianças. As causas da morte em adultos são fraqueza muscular grave e progressiva, disfagia, diminuição da nutrição, pneumonia por aspiração ou insuficiência respiratória devido a infecções pulmonares. A poliomiosite é mais grave e resistente ao tratamento na presença de dano ao coração e aos pulmões. A morte em crianças pode ocorrer devido a vasculite intestinal. O prognóstico geral da doença também é determinado pela presença de neoplasias malignas.