Médico especialista do artigo

Novas publicações
Síndrome de Angelman em crianças e adultos
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.

Existem várias doenças para as quais expressões como "cuide-se e não ficará doente" soam, no mínimo, ridículas. São patologias nas quais algumas anormalidades mentais e físicas são inerentes ao corpo da criança antes mesmo do nascimento, mas os pais não são culpados por isso. Essas doenças são causadas por mutações ou anormalidades nos conjuntos de cromossomos e são chamadas de cromossômicas ou genéticas. Síndrome de Angelman, síndrome de Down, síndrome de Patau, síndrome de Edwards, síndrome de Turner, síndrome de Prader-Willi - estas são apenas algumas das doenças genéticas de uma lista bastante razoável.
Síndrome do Homem Feliz
Desta vez, falaremos sobre a patologia que leva o nome do pediatra inglês Harry Angelman, que levantou a questão pela primeira vez em 1965, após ter encontrado três crianças incomuns em seu consultório no dia anterior, unidas por sintomas peculiares em comum. O médico chamou essas crianças de "crianças-bonecas" e escreveu um artigo sobre elas, inicialmente intitulado "Crianças-marionetes". O próprio artigo e seu título foram escritos sob a inspiração de uma pintura vista em um dos museus de Verona. A pintura retratava um menino rindo e era chamada de "O Menino Marionete". A associação da criança retratada na pintura com as três crianças que Angelman encontrou em seu consultório levou o pediatra a agrupar as crianças em um único grupo devido à doença que apresentavam.
Não é surpreendente que as crianças mencionadas no artigo não tenham sido notadas por outros médicos. Afinal, à primeira vista, parecia que elas tinham doenças completamente diferentes, tão diferente era o quadro clínico geral da doença em três casos distintos. Talvez a "nova" patologia cromossômica tivesse interessado outros cientistas, mas naquela época a genética ainda não estava suficientemente desenvolvida para confirmar a hipótese do médico inglês. Portanto, após certo interesse, o artigo foi deixado de lado por um longo tempo.
A próxima menção à síndrome de Angelman, que é como o artigo do pediatra inglês G. Angelman passou a ser chamado, remonta ao início dos anos 80 do século XX. E somente em 1987 foi possível descobrir a razão pela qual uma pequena parte das crianças nasce com tais anomalias que, vistas de fora, parecem estar constantemente sorrindo e felizes. Na verdade, isso não é verdade, e o sorriso é apenas uma careta, por trás da qual se esconde uma alma humana infeliz e a dor dos pais.
Epidemiologia
Segundo as estatísticas, uma mutação cromossômica em uma criança pode se desenvolver tanto na presença de mutações semelhantes nos pais quanto na ausência delas. Não há uma natureza hereditária clara da síndrome de Angelman (SA), mas a probabilidade de desenvolver a patologia em pais com mutações cromossômicas é bastante alta.
Também é interessante que, se uma família já tem uma criança com SA, há uma chance de um por cento de ter um segundo filho com o mesmo transtorno, mesmo que os pais sejam saudáveis.
Ainda não há estatísticas exatas sobre o número de pacientes com a síndrome de Angelman. Talvez a razão seja a variedade de sintomas, que podem ocorrer em uma determinada composição ou não ocorrer por um longo período. Estima-se que a prevalência da doença seja de 1 criança a cada 20.000 recém-nascidos. Mas esse número é muito aproximado.
Causas Síndrome de Angelman
Síndrome de Angelman é o nome médico para uma patologia cromossômica, mas está longe de ser a única. As pessoas chamam essa doença de síndrome da criança-boneca, síndrome do fantoche feliz, síndrome de Petrushka e síndrome da boneca risonha. As pessoas inventam todos os tipos de nomes (às vezes até ofensivos para os próprios pacientes e seus pais), mas uma doença é uma doença, não importa o quão engraçada ela pareça e não importa quais sejam as razões.
E as razões para o desenvolvimento da síndrome de Angelman, como muitas outras patologias genéticas, em todos os casos são distúrbios na estrutura de um dos cromossomos ou do conjunto cromossômico como um todo. Mas, no nosso caso, todo o problema está no cromossomo 15, transmitido pela mãe. Ou seja, o cromossomo paterno, neste caso, não apresenta anormalidades, mas o feminino sofre certas mutações.
De acordo com o tipo de anormalidade cromossômica, a síndrome de Angelman é classificada como uma mutação cromossômica. Tais mutações são consideradas:
- Uma deleção (ausência de uma seção de um cromossomo contendo um determinado conjunto de genes; se um dos genes estiver faltando, estamos falando de uma microdeleção), que é o resultado de duas quebras e uma reunião, quando uma seção do cromossomo original é perdida.
- Duplicação (presença de uma seção extra em um cromossomo que é uma cópia de um existente), que na maioria dos casos leva à morte de uma pessoa e, menos frequentemente, à infertilidade.
- Inversão (reversão de uma das seções do cromossomo em 180 graus, ou seja, na direção oposta, e então os genes nele estão localizados na ordem oposta), quando as extremidades quebradas do cromossomo são conectadas em uma ordem diferente da original.
- Inserção (se parte do material genético de um cromossomo estiver fora do lugar),
- translocação (se uma determinada seção de um cromossomo estiver ligada a outro cromossomo; tal mutação pode ser mútua sem perda de seções).
Ao receber um cromossomo mutado de uma mãe desavisada, a criança está fadada a nascer com anormalidades. A causa mais comum da síndrome de Angelman ainda é considerada uma deleção do 15º cromossomo materno, quando uma pequena parte dele está ausente. Mutações menos comuns na síndrome da "boneca que ri" são consideradas:
- translocação,
- dissomia unipaternal (se a criança recebeu um par de cromossomos do pai, o cromossomo materno está ausente),
- mutação de genes no DNA, que são tanto o principal material de construção (genético) quanto as instruções para seu uso correto (em particular, mutação do gene ube3a no cromossomo materno).
A presença de uma dessas mutações nos pais é um fator de risco para o desenvolvimento da síndrome de Angelman em crianças. Mas não apenas as mutações cromossômicas, mas também as genômicas (que estão associadas a uma alteração quantitativa nos conjuntos cromossômicos e são mais comuns do que as cromossômicas) podem provocar o desenvolvimento da doença em uma criança. Mutações genômicas comuns incluem trissomia cromossômica (se o conjunto cromossômico de uma pessoa tiver mais de 46 cromossomos).
Para que uma patologia apareça em uma criança, não é necessário que os pais tenham anormalidades cromossômicas. No entanto, existe uma certa porcentagem de pacientes cuja doença é hereditária.
Patogênese
Vamos nos aprofundar um pouco mais na biologia, ou mais precisamente, na genética. A informação genética de cada organismo humano está contida em 23 pares de cromossomos. Um cromossomo de cada par é transmitido à criança pelo pai, o outro pela mãe. Todos os pares de cromossomos diferem em forma e tamanho e carregam determinadas informações. Assim, o 23º par de cromossomos (cromossomos X e Y) é responsável pela formação das características sexuais do bebê (XX - menina, XY - menino, enquanto o cromossomo Y só pode ser recebido pela criança do pai).
Idealmente, uma criança recebe 46 cromossomos dos pais, que formam suas características genéticas, predeterminando-a como indivíduo. Um número maior de cromossomos é chamado de trissomia e é considerado um desvio da norma. Por exemplo, a presença do cromossomo 47 no conjunto cromossômico (cariótipo, que determina a espécie e as características individuais) causa a síndrome de Down.
Se os cromossomos forem corados com um corante especial, ao microscópio, é possível observar faixas de diferentes tonalidades ao longo de cada um deles. Dentro de cada faixa, há um grande número de genes. Todas essas faixas são numeradas pelos cientistas e têm uma localização fixa. A ausência de uma das faixas é considerada um desvio da norma. Na síndrome de Angelman, pode-se observar com muita frequência a ausência de segmentos do cromossomo materno no intervalo q11-q13, localizado no braço longo, cujo número de bases de DNA é de apenas cerca de 4 milhões.
O principal componente do cromossomo é considerado uma molécula de DNA incrivelmente longa, contendo milhares de genes e dezenas e centenas de milhões de bases nitrogenadas. Assim, o cromossomo 15, responsável pelo desenvolvimento da síndrome de Angelman e de várias outras doenças, contém 1.200 genes e cerca de 100 milhões de bases. Quaisquer distúrbios na estrutura da molécula de DNA certamente afetarão a aparência e o desenvolvimento da futura criança.
A informação genética contida nos genes é convertida em proteína ou RNA. Esse processo é chamado de expressão gênica. Dessa forma, a informação genética recebida dos pais recebe forma e conteúdo, que são incorporados em seu herdeiro único, feminino ou masculino.
Há uma série de patologias com um tipo de herança não clássica, incluindo a síndrome de Angelman, na qual genes recebidos dos pais como parte de cromossomos pareados carregam uma marca única dos pais e se manifestam de maneiras diferentes.
Assim, a síndrome de Angelman é um exemplo notável de imprinting genômico, em que a expressão gênica no corpo da criança depende diretamente de qual genitor os alelos foram recebidos (diferentes formas de um gene, recebido do pai e da mãe, localizadas em seções idênticas de cromossomos pareados). Ou seja, apenas anomalias no cromossomo materno levam ao desenvolvimento da síndrome, enquanto mutações e distúrbios estruturais no cromossomo paterno causam patologias completamente diferentes.
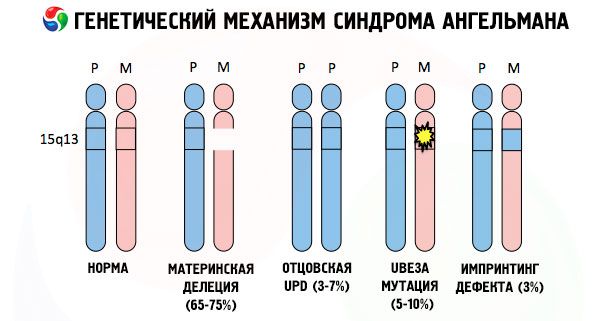
Nessa patologia, há ausência de certos genes no cromossomo materno ou perda/redução da atividade de genes individuais (na grande maioria dos casos, o gene ube3a, envolvido no metabolismo da ubiquitina, uma proteína que regula a degradação de outras proteínas). Como resultado, a criança é diagnosticada com anormalidades no desenvolvimento mental e deformidades físicas.
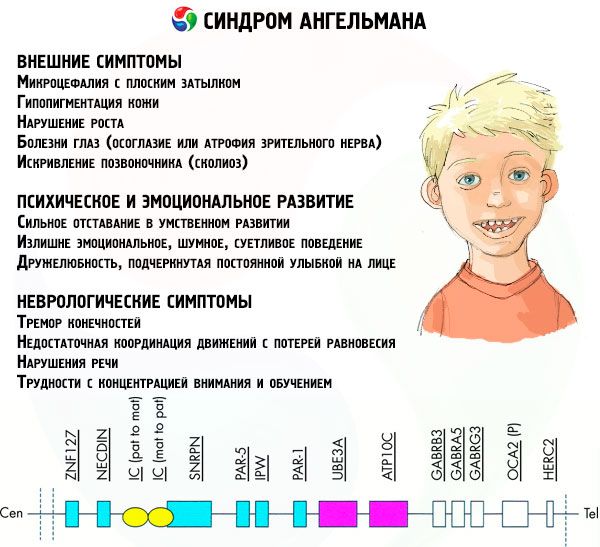
Sintomas Síndrome de Angelman
Os sintomas da síndrome de Angelman afetam vários aspectos da vida e do desenvolvimento da criança: físico, neurológico e mental. Com base nisso, podem ser identificados três grupos de sintomas que indicam o desenvolvimento desta patologia.
- Sintomas externos ou físicos:
- uma cabeça desproporcionalmente pequena em comparação com o corpo e os membros, que são de tamanho normal,
- boca muito larga,
- quase sempre há um sorriso no rosto (com a boca aberta),
- dentes esparsos,
- lábio superior estreito,
- língua larga e frequentemente projetada,
- maxilar inferior saliente,
- queixo pontudo,
- pele muito clara, frequentemente com pelos (albinismo, associado ao fato de o corpo não produzir o pigmento melanina),
- manchas escuras em pele clara (hipopigmentação devido à produção insuficiente de melanina)
- sintomas físicos ou externos: doenças oculares como estrabismo ou atrofia do nervo óptico,
- curvatura da coluna (escoliose),
- pernas rígidas (ao caminhar, a pessoa não dobra as pernas na altura dos joelhos devido à baixa mobilidade das articulações, daí a comparação com o andar de uma boneca).
- Sintomas relacionados ao desenvolvimento mental e emocional:
- retardo mental grave,
- comportamento excessivamente emocional, barulhento e exigente,
- palmas frequentes,
- simpatia expressa, enfatizada por um sorriso constante no rosto,
- risadas frequentes sem motivo.
- Sintomas neurológicos:
- tremor dos membros,
- coordenação insuficiente dos movimentos com perda de equilíbrio,
- diminuição do tônus muscular,
- vários distúrbios do sono,
- ataques histéricos frequentes na infância,
- distúrbios da fala (a criança começa a falar tarde, tem pouca capacidade de comunicação e fala arrastada),
- hiperatividade no contexto de aumento da excitabilidade,
- dificuldades de concentração e aprendizagem.
Mas este é um quadro generalizado da doença. De fato, o quadro clínico da síndrome de Angelman depende em grande parte do estágio de desenvolvimento da doença e do tipo de mutação cromossômica que causou a patologia. Isso significa que os sintomas da doença podem diferir significativamente em diferentes pacientes, o que por muito tempo não nos permitiu distinguir a patologia de outras com quadro clínico semelhante.
Dentre o total de sintomas, podemos destacar aqueles que são característicos de todos os pacientes sem exceção:
- retardo mental grave,
- comportamento inadequado (risos irracionais, aumento da excitabilidade, falta de concentração, estado de euforia),
- subdesenvolvimento das habilidades motoras,
- má coordenação dos movimentos, ataxia da marcha (ritmo irregular, oscilação de um lado para o outro, etc.), tremor dos membros.
- transtorno do desenvolvimento da fala com predominância de meios de comunicação não verbais.
Entre os sintomas encontrados pela grande maioria dos pacientes, podemos destacar:
- desproporção entre a cabeça e o corpo causada pelo atraso no desenvolvimento físico,
- em muitos pacientes, o formato do crânio é tal que o tamanho do cérebro permanece menor do que em pessoas saudáveis (microcefalia),
- crises epilépticas antes dos 3 anos de idade, com diminuição progressiva da força e da frequência em idade mais avançada,
- distorção dos parâmetros do EEG (flutuações e alta amplitude de ondas de baixa frequência).
Esses sintomas são bastante comuns, no entanto, 20% dos pacientes com síndrome de Angelman não os apresentam.
Ainda com menos frequência, é possível diagnosticar manifestações da doença como:
- estrabismo grave ou leve,
- controle deficiente do movimento da língua, resultando em pacientes que frequentemente colocam a língua para fora sem motivo,
- dificuldades de deglutição e sucção, especialmente em crianças pequenas,
- perturbação da pigmentação da pele e dos olhos,
- braços levantados ou dobrados ao caminhar,
- hiperreflexia,
- distúrbios do sono, especialmente na infância,
- salivação frequente,
- sede insaciável,
- movimentos de mastigação excessivamente ativos,
- hipersensibilidade ao calor,
- parte de trás da cabeça plana,
- maxilar inferior saliente,
- palmas lisas.
Uma porcentagem considerável de pacientes apresenta problemas com a micção, que não conseguem controlar bem, comprometimento da coordenação motora fina, o que gera dificuldades no autocuidado e na aprendizagem, e excesso de peso. Quase todos os pacientes atingem a puberdade mais tarde do que seus pares saudáveis.
Crianças com síndrome de Angelman percebem bem a fala oral e a compreendem, mas não querem participar de conversas, limitando sua fala a algumas dezenas de palavras necessárias ao dia a dia. No entanto, na idade adulta, esses pacientes parecem mais jovens do que seus pares sem patologias genéticas.
Muitos sintomas da síndrome de Angelman são inconstantes, de modo que o quadro clínico da doença muda significativamente com a idade. Convulsões e crises epilépticas tornam-se menos frequentes ou desaparecem completamente, o paciente fica menos excitável e o sono melhora.
Complicações e consequências
A síndrome de Angelman é uma patologia cromossômica grave, atualmente praticamente incurável, que priva os pacientes da oportunidade de viver uma vida normal. A vida de uma criança com SA depende em grande parte do tipo de anormalidade cromossômica.
A duplicação de um segmento cromossômico é incompatível com a vida na maioria dos casos. E mesmo que esses pacientes não morram na infância e cheguem à puberdade, não têm chance de ter filhos.
A deleção ou ausência de parte dos genes que ocorre com mais frequência na síndrome de Angelman é um obstáculo para a criança aprender a andar e falar. Essas crianças apresentam uma forma mais grave de retardo mental, e as crises epilépticas ocorrem com mais frequência, e sua intensidade é muito maior do que em pacientes com outras anormalidades cromossômicas.
Se houver apenas uma mutação de um gene, com a devida atenção e abordagem, a criança pode aprender noções básicas de autocuidado, comunicação e interação em grupo, embora ainda fique atrás de seus colegas no desenvolvimento.
Para crianças com Síndrome de Angelman, que são gentis por natureza, o mais importante é o amor e a atenção dos pais. Somente neste caso a educação da criança dará frutos, mesmo que pequenos. É claro que pacientes com Síndrome de Angelman não poderão estudar em uma escola regular. Eles precisam de aulas especiais, onde as crianças primeiro aprendam a se concentrar e, em seguida, gradualmente, aprendam o básico do conhecimento escolar.
Diagnósticos Síndrome de Angelman
A síndrome de Angelman é uma patologia congênita do desenvolvimento. Mas, devido a certas circunstâncias, muitas vezes é impossível diagnosticá-la na primeira infância. Isso se deve à inespecificidade e à fraca expressão dos sintomas em bebês e crianças menores de 3 anos. E a prevalência da doença em nosso país não é tão grande que os médicos tenham aprendido a reconhecê-la entre seus pares.
A síndrome de Angelman em bebês pode se manifestar como diminuição do tônus muscular, que se manifesta em problemas de alimentação (fraqueza do reflexo de sucção e deglutição) e, posteriormente, dificuldades em aprender a andar (essas crianças começam a andar muito mais tarde). Esses sintomas são os primeiros sinais de uma anormalidade no desenvolvimento do bebê, que pode estar associada a uma anormalidade cromossômica. Somente a análise genética pode confirmar essa suposição.
Atenção especial é dada às crianças cujos pais apresentam diversas doenças genômicas ou cromossômicas. Afinal, a doença pode não se manifestar a princípio e, se a patologia for detectada a tempo, com o início de um trabalho intensivo com a criança, é possível alcançar um sucesso significativamente maior na aprendizagem, retardando a progressão da doença.
Caso os pais apresentem diversas anomalias cromossômicas, a análise genética é realizada antes mesmo do nascimento do bebê, já que a SA é uma das patologias que podem ser detectadas ainda na fase embrionária.
A coleta de material para pesquisa genética pode ser realizada de duas maneiras:
- invasivo (com uma certa percentagem de risco, uma vez que é necessário penetrar no útero para recolher uma amostra de líquido amniótico),
- não invasivo (análise do DNA do bebê a partir do sangue da mãe).
Em seguida são realizados os seguintes estudos:
- hibridização fluorescente in situ (método FISH) – ligação de uma sonda de DNA marcada com um corante especial ao DNA em estudo, seguida de exame ao microscópio.
- análise de mutações no gene ube3a e genes impressos,
- Análise de metilação de DNA usando métodos especiais usados em genética.
Os testes genéticos fornecem informações bastante precisas em caso de anomalias cromossômicas, o que significa que os futuros pais sabem com antecedência o que esperar. No entanto, há exceções. Em um determinado grupo de pacientes, na presença de todos os sintomas indicativos de patologia, os resultados dos testes permanecem normais. Ou seja, a patologia só pode ser identificada observando-se atentamente a criança desde a primeira infância: como ela se alimenta, quando começou a andar e a falar, se dobra as pernas ao caminhar, etc.
Além do método FISH, entre os métodos instrumentais de diagnóstico da síndrome de Angelman, podemos destacar a tomografia computadorizada (TC ou RNM), que ajuda a determinar a condição e o tamanho do cérebro, e o eletroencefalograma (EEG), que mostra como funcionam as partes individuais do cérebro.
Os médicos geralmente fazem o diagnóstico final entre 3 e 7 anos de idade, quando o paciente já apresenta a maioria dos sintomas e a dinâmica do desenvolvimento da doença é visível.
Quais testes são necessários?
Diagnóstico diferencial
A síndrome de Angelman é uma patologia genética que praticamente não apresenta manifestações específicas. A maioria dos sintomas pode indicar tanto a síndrome de Asperger quanto outras patologias genéticas.
O diagnóstico diferencial da síndrome de Angelman é realizado com as seguintes patologias:
- Síndrome de Pitt-Hopkins (os pacientes são caracterizados por retardo mental, caráter alegre, sorriso, boca bastante grande e larga, sendo observada microcefalia). A diferença são crises de hiperventilação e retenção da respiração durante a vigília.
- Síndrome de Christianson (os pacientes são pessoas com retardo mental, de disposição alegre, incapazes de falar, caracterizadas por microcefalia, ataxia, convulsões, movimentos musculares involuntários).
- Síndrome de Mowat-Wilson (sintomas: retardo mental, crises epilépticas, queixo pontudo, boca aberta, expressão feliz no rosto, microcefalia). Distinção: grande distância entre os olhos, olhos puxados para dentro, ponta do nariz arredondada, aurícula voltada para trás.
- Síndrome de Kabuki (caracterizada por retardo mental leve a moderado, problemas de fala e motores, fraqueza muscular, crises epilépticas, microcefalia, longos intervalos entre coceiras e coordenação prejudicada). Caracterizada por sobrancelhas arqueadas, porção lateral evertida da pálpebra inferior, olhos bem separados, fissuras palpebrais longas com cílios longos e espessos.
- Síndrome de Rett (diferenciação da Síndrome de Rett em mulheres). Sintomas: atraso no desenvolvimento da fala, convulsões, microcefalia. A diferença é que não há expressão facial feliz, mas sim crises de apneia e apraxia, que progridem com o tempo.
- Síndrome de retardo mental autossômico recessivo 38 (sintomas: retardo mental acentuado com atrasos nas habilidades motoras e na fala, fraqueza muscular, problemas de alimentação na infância, impulsividade). Característica distintiva é a coloração azulada da íris.
- Síndrome de duplicação do gene MECP 2 (diferenciação da SA em homens). Sintomas: retardo mental grave, fraqueza muscular desde a infância, problemas de fala ou ausência de fala, epilepsia. Distinções: miopatia progressiva, infecções recorrentes constantes.
- Síndrome de Kleefstra (sintomas: problemas de fala e pensamento, fraqueza muscular, distúrbios do sono, falta de atenção, boca aberta, hiperatividade, convulsões, ataxia, distúrbios de equilíbrio). Características distintivas: rosto achatado, nariz curto e arrebitado, olhos bem separados, lábio inferior grande e evertido, explosões de agressividade.
- Síndrome de Smith-Magenis (caracterizada por convulsões, problemas de sono e distúrbios do desenvolvimento intelectual e motor). As características distintivas incluem rosto largo e achatado e testa proeminente.
- Síndrome de Koolen-de Vries (retardo mental leve a moderado, fraqueza muscular, convulsões, simpatia). Características distintivas: rosto longo com testa alta, orelhas proeminentes, olhos puxados, alta mobilidade articular, defeitos cardíacos congênitos.
- Síndrome de Phelan-McDermid (sintomas: retardo mental, distúrbios da fala ou ausência da fala). Características: mãos grandes com músculos desenvolvidos, fraqueza muscular congênita, sudorese fraca.
Patologias como deficiência de succinato de adenila, síndrome de retardo mental autossômico recessivo 1, síndrome de duplicação do cromossomo 2q23.1, síndromes de haploinsuficiência dos genes FOXG1, STXBP1 ou MEF2C e algumas outras podem “se gabar” de sintomas semelhantes à síndrome de Angelman.
A tarefa do médico é fazer um diagnóstico preciso, diferenciando a síndrome de Angelman de patologias com sintomas semelhantes, e prescrever um tratamento eficaz e relevante para o estágio diagnosticado da doença.
Quem contactar?
Tratamento Síndrome de Angelman
A síndrome de Angelman é uma das patologias para as quais a medicina ainda busca um tratamento eficaz. O tratamento etiológico da doença está em fase de desenvolvimento, com diversos métodos e meios, muitos dos quais ainda não testados em humanos. Isso significa que, por enquanto, os médicos precisam se limitar à terapia sintomática, o que ajuda a aliviar de alguma forma a situação nada invejável de crianças e adultos com síndrome de marionete, que sofrem de crises epilépticas, salivação excessiva, hipotensão e distúrbios do sono.
Assim, é possível reduzir a frequência e a intensidade das crises epilépticas com a ajuda de um anticonvulsivante adequadamente selecionado. Mas a dificuldade reside no fato de que as crises em pacientes com SA diferem das crises epilépticas comuns, pois são caracterizadas por vários tipos de crises, o que significa que a condição pode ser aliviada com a administração de vários medicamentos simultaneamente.
Os anticonvulsivantes mais populares para tratar a síndrome de Angelman são: ácido valproico, topiramato, lamotrigina, levetiracetam, clonazepam e medicamentos à base deles. Medicamentos à base de carmazepina, fenitoína, fenobarbital e etossuximida são menos comumente utilizados, pois alguns deles podem provocar um efeito paradoxal, que consiste no fortalecimento e aumento da frequência das crises epilépticas. Isso ocorre se o medicamento for usado como parte de monoterapia.
Para tratar a salivação excessiva, geralmente são utilizados dois métodos: medicamentoso (medicamentos que suprimem a produção de saliva) e cirúrgico, que envolve o reimplante dos ductos salivares. Mas, no caso da salivação excessiva, esses métodos são considerados ineficazes e a questão permanece em aberto. Pais e cuidadores desses pacientes devem prestar atenção especial a essa questão, visto que os próprios pacientes geralmente não controlam a salivação e alguns simplesmente não conseguem cuidar de si mesmos.
Outro problema é a curta duração do sono. Muitas vezes, crianças com síndrome de Angelman dormem no máximo 5 horas, o que tem um impacto negativo no funcionamento de todo o corpo. Crianças facilmente excitáveis e ativas, que adoram brincadeiras e comunicação (mesmo que tentem se limitar a métodos não verbais), sentem-se visivelmente cansadas durante o dia. Para ter um bom descanso, o corpo precisa de um sono profundo e completo, mas esse é justamente o problema.
Aparentemente, medicamentos sedativos (fenotiazinas e antipsicóticos atípicos) que acalmam o sistema nervoso deveriam ser suficientes para melhorar o sono em pacientes excitáveis. Mas, no caso da Síndrome de Asperger, o uso desses medicamentos está repleto de efeitos negativos. Portanto, os médicos ainda preferem pílulas para dormir leves, como a Melatonina (um medicamento hormonal natural baseado no hormônio do sono), que é administrado aos pacientes uma hora antes de dormir, na quantidade de 1 comprimido, e a Difenidramina. A frequência de administração e a dosagem são determinadas pelo médico, dependendo da condição e da idade do paciente.
Às vezes, pacientes com síndrome de Angelman apresentam problemas de digestão e fezes. Você pode melhorar suas fezes com laxantes (de preferência à base de ervas).
Ou você pode abordar o problema de forma diferente, como fizeram os médicos americanos, com base em alguns métodos de tratamento do autismo, porque muitos sintomas característicos da Síndrome de Asperger também são característicos do autismo (impulsividade, movimentos involuntários, ações repetitivas, déficit de atenção, problemas de comunicação, etc.). Observou-se que a introdução do hormônio secretina, que normaliza a digestão e as fezes, tem um efeito positivo na atenção dos pacientes, e a ocitocina ajuda a melhorar as habilidades cognitivas e a memória da criança, além de corrigir o comportamento.
É verdade que os hormônios por si só não são suficientes, especialmente quando se trata de crianças. Na síndrome de Angelman, são indicados terapia comportamental, acompanhamento com psicólogo e fonoaudiólogo (ensino de métodos de comunicação não verbal e linguagem de sinais). A educação dessas crianças deve ser baseada em um programa individual com a participação de professores especialmente treinados, psicólogo e pais. Infelizmente, isso não é possível em todos os lugares, e as famílias acabam ficando sozinhas com o problema.
Como muitos pacientes jovens com EA sofrem de baixo tônus muscular e problemas articulares, muita atenção é dada à fisioterapia. Na maioria das vezes, os médicos recorrem ao uso de aplicações de parafina, eletroforese e terapia magnética.
Massagem tônica ativa e exercícios especiais de fisioterapia ajudarão a criança doente a se levantar e caminhar com confiança depois de um tempo. A hidroginástica é especialmente útil nesse sentido, sendo recomendada para SA em água fria. Ela aumenta o tônus muscular e ensina a criança a controlar o corpo e coordenar os movimentos.
Tratamento anticonvulsivante
O sintoma mais perigoso da síndrome de Angelman são convulsões semelhantes às da epilepsia. Esse sintoma é observado em 80% dos pacientes, o que significa que todos eles precisam receber tratamento anticonvulsivante eficaz.
O tratamento das crises epilépticas é realizado com o auxílio de vitaminas e anticonvulsivantes. Na síndrome de Angelman, acompanhada de síndrome convulsiva, vitaminas do complexo B, bem como as vitaminas C, D e E, serão úteis. No entanto, a automedicação com vitaminas neste caso é muito perigosa, pois a ingestão descontrolada de vitaminas pode reduzir a eficácia dos medicamentos antiepilépticos e provocar novas crises, mais graves e prolongadas.
A seleção dos medicamentos anticonvulsivantes e a prescrição da dosagem eficaz também devem ser feitas por um médico especialista. Ele também decidirá se um único medicamento será suficiente ou se o paciente precisará tomar dois ou mais medicamentos por um longo período.
Para a maioria dos pacientes, os médicos prescrevem medicamentos com ácido valpróico (ácido valpróico, Depakine, Convulex, Valparin, etc.), que previnem convulsões e melhoram o humor e o estado mental dos pacientes.
O ácido valproico está disponível na forma de comprimidos, xarope e soluções injetáveis. O medicamento mais popular é o Depakine, de liberação prolongada, em comprimidos e solução para administração intravenosa. A dosagem do medicamento é determinada individualmente pelo médico, dependendo do peso, idade e condição do paciente.
O medicamento é tomado durante as refeições, de 2 a 3 vezes ao dia. A dose média diária é de 20 a 30 mg por quilo de peso do paciente, sendo a máxima de 50 mg/kg por dia.
Contraindicações de uso. Não utilizar em caso de disfunção hepática e pancreática, diátese hemorrágica, hepatite, porfiria e hipersensibilidade ao medicamento.
Os efeitos colaterais incluem tremores nas mãos, distúrbios digestivos e nas fezes, e alterações no peso corporal.
O "topiramato" também é um medicamento de escolha para SA. É produzido em comprimidos e usado tanto como monoterapia quanto em combinação com outros medicamentos.
Modo de administração e dosagem. Tome os comprimidos por via oral, independentemente da ingestão de alimentos. A dose diária inicial para adultos é de 25 a 50 mg, para crianças - 0,5 a 1 mg/kg. A cada semana, a dosagem é aumentada de acordo com as instruções do médico.
O medicamento não deve ser tomado durante a gravidez e a lactação, bem como em caso de hipersensibilidade aos seus componentes. O medicamento apresenta diversos efeitos colaterais.
Medicamentos que um médico pode prescrever para a síndrome de Angelman: Clomazepam, Rivotril, Lamotrigina, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Medicina tradicional e homeopatia
A medicina tradicional, como os preparados homeopáticos, é obviamente relativamente segura, mas a eficácia desse tratamento para a síndrome de Angelman pode ser considerada controversa.
Embora o tratamento popular ainda possa ajudar em algumas coisas. Estamos falando de interromper as crises epilépticas. Nesse sentido, o tratamento com ervas pode ser bastante eficaz.
Um bom efeito é proporcionado por uma coleção medicinal à base de peônia, alcaçuz e lentilha-d'água (os componentes são tomados em quantidades iguais). As ervas precisam ser moídas até virar farinha. Após 2 semanas do início do uso, você poderá notar uma diminuição significativa na frequência das convulsões.
A decocção de lavanda (1 colher de chá por copo de água fervente) também é útil para cólicas. A mistura é fervida por 5 minutos e infundida por meia hora. O medicamento é tomado à noite por 14 dias.
Uma infusão aquosa (ou alcoólica) de erva-mãe é considerada eficaz para convulsões epilépticas.
Entre os preparados homeopáticos para a prevenção de convulsões na síndrome de Angelman, você pode usar medicamentos à base de camomila e erva-mãe, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum e Arsenicum album. No entanto, é importante lembrar que somente um médico homeopata pode prescrever doses eficazes e seguras de medicamentos em cada caso específico.
Prevenção
Como o leitor provavelmente já entendeu, a medicina ainda não é capaz de prevenir mutações genéticas e outras anormalidades cromossômicas, nem de corrigir a situação. Isso pode acontecer com qualquer pessoa, pois crianças com síndrome de Angelman nascem de pais saudáveis, e a genética, que atualmente é um dos ramos menos estudados da medicina, ainda não consegue explicar isso.
A única coisa que pode ser feita é adotar uma abordagem responsável em relação ao planejamento da gravidez, registrar-se e realizar os exames em tempo hábil. Mas, novamente, tal medida terá um caráter mais educativo do que preventivo, como qualquer exame. No entanto, os jovens pais saberão com antecedência para o que se preparar e, em caso de resposta positiva, decidirão se podem assumir a responsabilidade de criar um filho doente.
Previsão
O prognóstico da síndrome de Angelman depende da natureza da anomalia cromossômica e da rapidez de sua detecção. As mais afetadas são as crianças cujo cromossomo 15 contém "lacunas" nos genes (deleção). A probabilidade de esses pacientes andarem e falarem é extremamente baixa. Outros casos podem ser corrigidos com uma abordagem cuidadosa e amor ao seu filho.
Infelizmente, esses pacientes não poderão se tornar membros plenos da sociedade, apesar de estarem longe de ser estúpidos, pois compreendem a fala e seu significado. No entanto, terão problemas de comunicação pelo resto da vida. Os pacientes podem aprender a língua de sinais desde a infância, mas não podem ser forçados a se comunicar usando palavras. O vocabulário dos pacientes "falantes" é limitado ao mínimo de palavras usadas na vida cotidiana (5 a 15 palavras).
Quanto à expectativa de vida e à saúde geral dos pacientes com síndrome de Angelman, os números aqui oscilam em torno da média. Na idade adulta, os pacientes enfrentam principalmente problemas de saúde como escoliose e obesidade, que, com a abordagem correta ao tratamento, não representam risco de vida.