Médico especialista do artigo

Novas publicações
Raiva em crianças
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
A raiva, ou hidrofobia, é uma doença viral aguda transmitida pela mordida de um animal infectado, com danos ao sistema nervoso e desenvolvimento de encefalite grave com desfecho fatal.
Epidemiologia
Um flagelo da saúde pública desde a antiguidade, o vírus da raiva causa atualmente cerca de 59.000 mortes humanas por ano, quase todas transmitidas por mordeduras de cães. Isso tem um impacto econômico significativo nos países em desenvolvimento, particularmente na África e na Ásia, que podem suportar menos perdas. No entanto, apesar de sua taxa de mortalidade de quase 100%, a raiva canina é uma doença totalmente prevenível, e exemplos históricos de erradicação da raiva canina no mundo desenvolvido atestam isso. [ 1 ]
Causas raiva
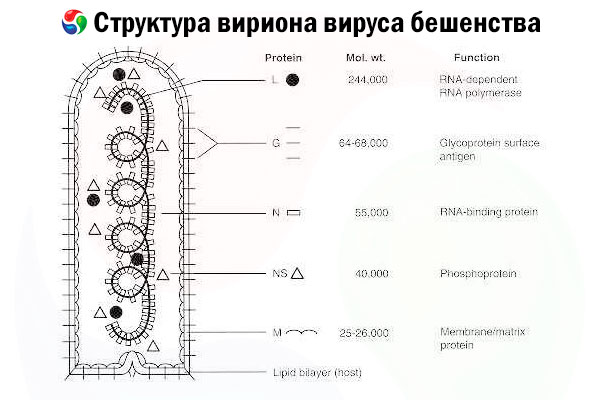
O agente causador é o vírus da raiva (RV), um vírus de RNA de fita negativa da família dos rabdovírus, com tamanho aproximado de 60 nm × 180 nm.
Consiste em um núcleo proteico interno, ou nucleocapsídeo, contendo ácido nucleico, e uma membrana externa, uma bicamada contendo lipídios, coberta por picos de glicoproteínas transmembrana. Possui uma estrutura genômica modular relativamente simples e codifica cinco proteínas estruturais:
- RNA polimerase dependente de RNA (L),
- nucleoproteína (N),
- proteína fosforilada (P),
- proteína da matriz (M) e
- glicoproteína de superfície externa (G).
As proteínas N, P e L, juntamente com o RNA genômico, formam o complexo ribonucleoproteico. G é o único antígeno de RV capaz de induzir a produção de anticorpos neutralizantes de RV, que são os principais efetores imunológicos contra a infecção letal por RV. Por outro lado, o complexo ribonucleoproteico demonstrou ser o principal antígeno de RV capaz de induzir células T CD4+, o que pode aumentar a produção de anticorpos neutralizantes de RV por meio do reconhecimento de antígenos intraestruturais.[ 2 ] O complexo ribonucleoproteico pode desempenhar um papel importante no estabelecimento da memória imunológica e da imunidade de longo prazo.[ 3 ]
 [
[ Classificação e tipos de antígenos
O gênero Lyssavirus inclui o vírus da raiva e vírus da raiva relacionados antigenicamente e geneticamente: os vírus Lagos, Mokola e Duvenhage, bem como dois subtipos putativos de lyssavírus de morcegos europeus. Estudos de proteção cruzada indicam que animais imunizados com vacinas antirrábicas tradicionais podem não estar totalmente protegidos quando desafiados com outros lyssavírus.
Os vírus da raiva podem ser classificados como fixos (adaptados por passagem em animais ou cultura de células) ou de rua (tipo selvagem). O uso de anticorpos monoclonais e sequenciamento genético para diferenciar os vírus da raiva de rua ajudou a identificar variantes virais originárias dos principais reservatórios de hospedeiros em todo o mundo e a sugerir prováveis fontes de exposição humana quando um histórico de mordida animal definitiva estava ausente no caso de um paciente.[ 8 ]
Patogênese
Os principais reservatórios e fontes de infecção entre animais selvagens são lobos, raposas, chacais, morcegos e, entre animais domésticos, cães e gatos, raramente, cavalos, gado, porcos, ratos, etc. A transmissão de pessoa para pessoa, embora possível, é extremamente rara. Esta é uma infecção zoonótica típica. As pessoas são infectadas com raiva principalmente por meio de cães.
Após um ser humano ser mordido por um animal doente, o vírus se multiplica no tecido muscular no local da picada e, em seguida, tendo atingido as extremidades dos nervos periféricos sensoriais, espalha-se centripetamente, atingindo os neurônios motores. O tempo que o vírus leva para se mover e o cérebro ser afetado depende do local da picada. Em caso de picadas graves na cabeça e no rosto, o vírus pode atingir o sistema nervoso central em 15 a 20 dias e, em caso de danos leves na pele do tronco e membros e, consequentemente, uma pequena dose do patógeno, o processo de movimentação do vírus para o sistema nervoso central pode ser retardado por vários meses ou até mesmo 1 a 1,5 anos. Uma vez atingido o sistema nervoso central, o vírus se fixa nos tecidos do cérebro e da medula espinhal, principalmente nos neurônios da medula oblonga, do corno de Amon e da base do cérebro. Na medula espinhal, os cornos posteriores são os mais afetados. Do sistema nervoso central, o vírus se move centrifugamente ao longo dos troncos nervosos até as glândulas salivares, onde se multiplica e é excretado com a saliva.
Conceitos na patogênese da raiva
O RV possui uma ampla gama de hospedeiros e pode infectar quase todos os mamíferos. Embora várias rotas de transmissão do RV tenham sido relatadas, a infecção natural ocorre mais comumente por meio de uma mordida. Além das mordidas, o consumo de carcaças infectadas por RV pode promover a infecção pelo vírus da raiva em raposas do Ártico, e o contato do RV com membranas mucosas foi considerado outra possível rota de transmissão. [ 9 ] Em algumas circunstâncias incomuns, como a liberação acidental de RV como aerossol em um laboratório ou RV como aerossol em cavernas habitadas por um grande número de morcegos, [ 10 ] a transmissão por aerossol pode ocorrer.
Ainda não está claro se as cepas de RV de rua e adaptadas a camundongos ou adaptadas à cultura de tecidos se replicam no local da inoculação antes de entrarem no SNC. Enquanto a infecção intramuscular experimental de hamsters ou guaxinins juvenis com RV de rua revelou a replicação do RV em células musculares estriadas antes do vírus invadir os axônios dos neurônios motores através das junções neuromusculares,[ 11 ],[ 12 ] a infecção intramuscular de camundongos com RV CVS-24 adaptado a camundongos mostrou que o RV migra diretamente para o SNC sem replicação prévia no local da inoculação.[ 13 ] Uma vez nos terminais dos axônios amielínicos, o RV é transportado retrógrada para o corpo celular.
Descobertas recentes sugerem que o transporte de vesículas axonais pode representar uma estratégia chave para o movimento de vírions de longa distância nos axônios. [ 14 ] Foi estimado que o RV migra dentro dos axônios a uma taxa de 3 mm/h. [ 15 ] A infecção então se espalha através de uma cadeia de neurônios conectados por junções sinápticas. No entanto, o mecanismo exato que promove a disseminação transsináptica ainda é desconhecido. Após infectar o cérebro, o vírus se espalha centrifugamente para o sistema nervoso periférico e autônomo em muitos órgãos periféricos. [ 16 ] No último estágio do ciclo de infecção, o RV migra para as glândulas salivares; após a replicação em células acinares mucogênicas, ele é liberado na saliva e está pronto para transmissão ao próximo hospedeiro. [ 17 ]
No que diz respeito à patologia induzida pelo vírus da raiva, a morte celular apoptótica foi proposta como um potencial mecanismo patogênico em modelos experimentais de raiva de camundongos infectados com uma cepa fixa de RV. [ 18 ] Um mecanismo patogênico que pode contribuir para a profunda disfunção do SNC característica da raiva pode ser a função neuronal prejudicada. A expressão genética demonstrou ser marcadamente reduzida em neurônios infectados por RV, resultando em uma supressão geral da síntese de proteínas, [ 19 ] e vários estudos mostraram neurotransmissão prejudicada após infecção por RV. Jiang demonstrou que a ligação de um antagonista do receptor de acetilcolina a homogeneizados de cérebro de rato infectados foi reduzida em comparação com os controles. [ 20 ] A liberação e a ligação prejudicadas de serotonina, um neurotransmissor envolvido no controle do ciclo do sono, percepção da dor e comportamento, também foram observadas no cérebro de rato infectado por RV. [ 21 ], [ 22 ] Além de afetar a neurotransmissão, a infecção do ventrículo direito também pode afetar os canais iônicos. As células de neuroblastoma de camundongo infectadas apresentam expressão funcional diminuída dos canais de sódio dependentes de voltagem, o que pode impedir potenciais de ação e, em última análise, levar ao comprometimento funcional. [ 23 ]
Além da ausência de lesões patológicas graves no SNC, a maioria dos casos de raiva humana não provoca uma resposta imune de 7 a 10 dias após o início dos sinais clínicos. Essas diferenças profundas entre a patogênese da raiva e a da maioria das outras infecções virais ou bacterianas do SNC são ainda mais apoiadas pelo fato de que a imunossupressão é ineficaz ou prejudicial ao resultado da raiva. [ 24 ] O baixo nível de resposta imune frequentemente observado em vítimas de raiva é intrigante porque não pode ser explicado pela baixa imunogenicidade dos antígenos do RV. Na verdade, a proteína G e do nucleocapsídeo do RV são antígenos potentes de células B e T quando administrados parenteralmente. [ 25 ] Uma possível explicação para o baixo grau de resposta imune contra o RV em humanos ou animais com raiva pode ser que a infecção do SNC pelo RV causa imunossupressão, [ 26 ] e foi proposto que o RV usa uma estratégia subversiva, incluindo a prevenção da apoptose e a destruição de células T invasoras. [ 27 ]
Cepas de RV atenuadas que foram adaptadas a células não neuronais diferem significativamente das cepas patogênicas de RV de rua em sua neuroinvasividade, que se refere à sua capacidade de invadir o SNC a partir de locais periféricos. Nesse sentido, cepas de RV adaptadas à cultura de tecidos não têm ou têm capacidade limitada de invadir o SNC a partir de locais periféricos, enquanto cepas de RV de rua ou cepas de RV adaptadas a camundongos, como CVS-24, são altamente invasivas. [ 28 ] Os principais fatores envolvidos na neuroinvasão de RV incluem captação viral, transporte axonal, disseminação transsináptica e taxa de replicação viral.
Até recentemente, nosso conhecimento sobre a patogênese do RV era limitado e se baseava principalmente em estudos descritivos de cepas de RV encontradas em ruas ou em infecções experimentais com cepas atenuadas adaptadas em laboratório. O advento da tecnologia de genética reversa nos permitiu identificar os elementos virais que determinam o fenótipo patogênico do RV e compreender melhor os mecanismos envolvidos na patogênese da raiva.
Identificação de elementos virais que controlam a aquisição, disseminação e replicação do vírus da raiva
- Elementos virais envolvidos na captura do vírus
A infecção por RV começa com a ligação do vírus a um receptor celular putativo. Embora várias moléculas de superfície de membrana tenham sido propostas como receptores de RV, incluindo o receptor nicotínico de acetilcolina,[ 29 ] a molécula de adesão de células neurais[ 30 ] e o receptor de neurotrofina de baixa afinidade p75 NTR,[ 31 ] ainda não está claro se essas moléculas realmente desempenham um papel no ciclo de vida do vírus da raiva. Nesse contexto, foi demonstrado recentemente que a interação RV G–p75 NTR não é necessária para a infecção por RV de neurônios primários.[ 32 ] Após a ligação ao receptor, o RV é internalizado por meio de endocitose adsortiva ou mediada por receptor. [ 33 ] O ambiente de baixo pH dentro do compartimento endosomal induz então mudanças conformacionais no RV G que desencadeiam a fusão da membrana viral com a membrana endosomal, liberando assim o RNP no citoplasma. [ 34 ] Para vírus, RV G desempenha um papel crítico na captação viral, provavelmente por meio de interações com receptores celulares putativos que facilitam a captação rápida. A este respeito, foi demonstrado que a patogenicidade de cepas de RV adaptadas à cultura de tecidos (por exemplo, ERA, HEP e CVS-11) se correlaciona com a presença de um determinante localizado no sítio antigênico III da proteína G. [ 35 ] Uma mutação Arg → Gln na posição 333 neste sítio antigênico da proteína ERA G resultou em um atraso de sete vezes na internalização da variante RV Gln333 em comparação com a variante selvagem. A mutação Asn194→Lys194 no RV G, que explica o ressurgimento do fenótipo patogênico, foi associada a uma diminuição significativa no tempo de internalização.[ 36 ] Além disso, experimentos com RVs quiméricos mostraram que o tempo necessário para a internalização dos vírions do RV foi significativamente aumentado e a patogenicidade foi fortemente reduzida após a substituição do gene G da cepa SB RV altamente patogênica, que foi derivada de um clone de cDNA da cepa RV-18 associada a morcegos derivada da prata,[ 37 ] com a da cepa SN altamente atenuada, que foi isolada de um clone de cDNA da cepa da vacina SAD B19 RV.[ 38 ] Juntos, esses dados apoiam a noção de que a cinética da absorção do vírus, que é uma função do RV G, é um determinante importante da patogenicidade do RV.
- Elementos virais envolvidos na propagação e transmissão de vírus
Uma propriedade única do vírus da raiva é sua capacidade de se espalhar de célula para célula. A observação de que a variante Gln333 ERA perde a atividade de fusão célula-célula dependente de pH in vitro [ 39 ] e exibe uma capacidade muito reduzida de se espalhar de célula para célula [ 40 ] sugere que o RV G também desempenha um papel fundamental na disseminação de célula para célula e, portanto, na transmissão do vírus, provavelmente por meio de sua atividade fusiogênica. Essa possibilidade é ainda mais apoiada pela descoberta de que a taxa de disseminação do SPBNGAK reversível de RV patogênico é quase duas vezes maior do que a determinada para a variante SPBNGA não patogênica. Curiosamente, a mutação Asn 194 → Lys 194 em G SPBNGAK causou uma mudança no limite de pH para fusão de membrana para um pH mais alto, apoiando a hipótese de que um limite de pH mais alto para fusão de membrana está associado ao aumento da disseminação do vírus. [ 41 ]
Estudos de indicadores transneuronais de infecção por RV em ratos [ 42 ] e macacos rhesus [ 43 ] mostraram que o vírus da raiva migra exclusivamente em uma direção retrógrada nos axônios. Embora várias proteínas do RV estejam envolvidas em mecanismos de transporte neuronal, o RV G parece desempenhar um papel predominante na disseminação transneuronal da infecção por RV. Por exemplo, enquanto a infecção periférica com o vírus da anemia infecciosa equina (EIAV) pseudotipado com RV G resulta em transferência viral para a medula espinhal, o mesmo EIAV pseudotipado com o vírus da estomatite vesicular G não conseguiu entrar no sistema nervoso. [ 44 ] Além disso, a disseminação viral do mutante ERA G Arg 333 → Gln 333 no SNC foi fortemente reduzida em comparação com o mutante do tipo selvagem, sugerindo ainda uma função do RV G intacto na disseminação transsináptica. No entanto, a evidência mais convincente para um papel importante do RV G no transporte trans-sináptico vem da infecção intracraniana de camundongos com um vírus RV recombinante deficiente em G, que mostrou que a infecção permaneceu restrita aos neurônios no local da inoculação sem qualquer evidência de disseminação para neurônios secundários. [ 45 ] No entanto, é provável que, além do RV G, o RV M também desempenhe um papel na disseminação do vírus e, portanto, no transporte trans-sináptico. Nesse sentido, foi demonstrado que a disseminação da variante quimérica SN-BMBG RV, que contém M e G do SB altamente patogênico, foi significativamente maior do que a disseminação da variante quimérica SN-BG ou SN-BM, que contêm G e M do SB, respectivamente, sugerindo que a interação ótima de M com G pode desempenhar um papel importante na disseminação do vírus de célula para célula. [ 46 ] Uma vez que o RV M suporta a brotação do vírus, [ 47 ] é provável que a propagação mais eficiente da variante quimérica RV SN-BMBG seja devido à brotação ideal do vírus na membrana pós-sináptica.
Estudos recentes demonstraram que a interação entre o RV P e a cadeia leve da dineína liga o RV RNP ao sistema de transporte da célula hospedeira, facilitando assim o transporte axonal retrógrado do vírus.[ 48 ],[ 49 ] No entanto, a infecção periférica de camundongos adultos mostrou que a exclusão do domínio de ligação LC8 do RV P não impede a entrada do vírus no SNC, sugerindo que a proteína RV não está diretamente envolvida na disseminação axonal retrógrada do RV.[ 50 ]
- Elementos virais que controlam a replicação viral
Ao contrário de muitos outros vírus, como o vírus da gripe, a patogenicidade do RV é inversamente proporcional à taxa de síntese de RNA viral e produção de partículas virais infecciosas. A comparação dos níveis de mRNA viral e RNA genômico produzidos por diferentes vírus quiméricos sugere que a transcrição e replicação do RNA viral são reguladas por múltiplos fatores, incluindo RV M, que foi identificado como um fator trans-atuante que medeia a mudança de altos níveis iniciais de síntese de mRNA para síntese de RNA genômico. [ 51 ] Além disso, M de todos os rabdovírus é capaz de desligar a expressão do gene viral ligando-se ao RNP, resultando na formação de uma estrutura semelhante a uma espinha dorsal altamente condensada que é incapaz de suportar a síntese de RNA.
Para identificar outros elementos virais que controlam a patogenicidade por meio da regulação da replicação viral, as sequências terminais 5' da cepa SB altamente patogênica foram substituídas gradualmente por sequências da cepa vacinal SN altamente atenuada, resultando nos vírus recombinantes SB2 (sequência terminal [TS] + L), SB3 (TS + L + pseudogene [Ψ]), SB4 (TS + L + Ψ + G) e SB5 (TS + L + Ψ + G + M). A infecção intramuscular com os vírus parentais SB e SN e os RVs quiméricos SB2, SB3, SB4 e SB5 provocou as maiores taxas de mortalidade em camundongos infectados com SB e nenhuma morbidade ou mortalidade em camundongos infectados com SN. A substituição de TS, L e SB pelos elementos correspondentes da SN resultou em uma redução modesta na morbidade e mortalidade, e uma troca adicional de G ou G mais M reduziu fortemente ou aboliu completamente a patogenicidade viral.
A caracterização fenotípica desses RVs selvagens e quiméricos em cultura de tecidos revelou que a patogenicidade de um determinado RV é inversamente correlacionada com sua capacidade de se replicar em células neuronais. Embora o SB tenha se replicado em níveis quase 1.000 vezes menores que o SN, e a substituição dos níveis de TS, L e SB por SN tenha tido pouco efeito na cinética de crescimento viral, a substituição adicional do G ou G mais M do SB pelos genes SN correspondentes resultou em um aumento de 1 logaritmo na produção viral, sugerindo que a cinética de replicação do RNA viral, bem como a produção de partículas virais, são amplamente controladas pela proteína G do RV. Essa conclusão é corroborada por dados obtidos com variantes G do RV que diferem em um aminoácido em suas proteínas G. A variante patogênica do vírus da raiva SPBNGAK 194 produziu um título viral em células NA que foi 1 log menor do que o produzido pela variante não patogênica SPBNGAN 194, e a análise de PCR em tempo real mostrou que as taxas de transcrição e replicação do RNA viral em células NA infectadas com SPBNGAK foram 5 e 10 vezes maiores do que em células NA infectadas com SPBNGAK. [ 52 ] Mais evidências de uma correlação inversa entre patogenicidade e a taxa de síntese de RNA viral e produção de partículas virais foram fornecidas por camundongos infectados com vírus recombinantes quiméricos nos quais os genes G e M da cepa SN atenuada foram substituídos por aqueles da cepa SB altamente patogênica. Esses experimentos revelaram um aumento significativo na patogenicidade da cepa SN parental carregando RV G sobre a cepa SB patogênica. A patogenicidade foi ainda mais aumentada quando G e M de SB foram introduzidos em SN.
A substituição de G ou M, ou ambos, em SN pelos genes correspondentes de SB foi associada a uma diminuição significativa na taxa de produção de partículas virais, bem como na taxa de síntese de RNA viral. Esses dados indicam que tanto G quanto M desempenham papéis importantes na patogênese do RV, regulando a replicação viral. A descoberta de que a substituição de G ou G mais M em SN por G ou G mais M de SB resulta em uma diminuição moderada a forte na transcrição e replicação do RNA viral, respectivamente, enquanto a substituição de M sozinho em SN por M de SB resulta em um forte aumento na transcrição e replicação do RNA viral, indica que o RV G também tem uma importante função reguladora na transcrição/replicação do RNA viral, sozinho ou por meio da interação com a proteína M. O mecanismo pelo qual o gene RV G controla a síntese de RNA viral é desconhecido. Certas sequências de nucleotídeos dentro dos genes RV G, como aquelas que incluem os códons para Arg333 e Lys 194, foram identificadas como alvos para miRNAs celulares. Foi demonstrado que o reconhecimento do alvo por miRNAs celulares pode resultar em regulação positiva ou negativa da replicação viral. [ 53 ] As substituições de Arg 333 → Glu 333 ou Lys 194 → Ser 194 na sequência do gene G do RV resultam na abolição das sequências alvo de miRNA, o que por sua vez está associado a um aumento significativo na taxa de síntese de RNA viral [Faber M, Thomas Jefferson University, PA, EUA, dados não publicados], sugerindo que os miRNAs celulares do hospedeiro também desempenham um papel importante na regulação da replicação do RV, como foi demonstrado para outros vírus de RNA, incluindo o vírus da estomatite vesicular e o HCV. [ 54 ], [ 55 ]
A regulação da replicação viral parece ser um dos mecanismos importantes envolvidos na patogênese do RV. Para escapar da resposta imune e preservar a integridade da rede neuronal, cepas patogênicas de RV, mas não cepas atenuadas, podem regular sua taxa de crescimento. Uma menor taxa de replicação provavelmente beneficia as cepas patogênicas de RV, preservando a estrutura neuronal que esses vírus utilizam para atingir o SNC. Outra explicação para a menor taxa de replicação do RV patogênico é que, para escapar da detecção precoce pelo sistema imunológico do hospedeiro, o vírus mantém níveis mínimos de expressão de seus antígenos.
Relação entre expressão de RV G, apoptose e patogenicidade
É bem conhecido que as cepas do vírus da raiva de rua que são significativamente mais patogênicas do que as cepas adaptadas à cultura de tecidos expressam níveis muito limitados de G e não induzem apoptose até o final do ciclo infeccioso, sugerindo que a patogenicidade de uma cepa de vírus específica é inversamente correlacionada com a expressão de G do RV e a capacidade de induzir apoptose. [ 56 ] Evidências diretas de uma correlação entre o nível de expressão de G e a extensão da apoptose foram obtidas com o RV recombinante SPBNGA-GA, que carregava dois genes G idênticos e superexpressava RV G. [ 57 ] Estudos morfológicos de culturas neuronais infectadas com este RV recombinante mostraram que a morte celular aumentou significativamente em paralelo com a superexpressão de G do RV e que a apoptose é o principal mecanismo envolvido na morte mediada por G do RV. Em particular, a diminuição na coloração de F-actina após a infecção por SPBNGA-GA é consistente com a despolimerização induzida por apoptose de filamentos de actina. Além disso, o número de núcleos TUNEL-positivos em neurônios infectados por SPBNGA-GA foi significativamente aumentado em comparação com aqueles em neurônios não infectados e infectados por SPBNGA. No entanto, o mecanismo pelo qual o gene RV G medeia o processo de sinalização apoptótica permanece em grande parte desconhecido. Foi sugerido que a expressão de RV G acima de um certo limite interrompe gravemente a membrana celular. É altamente provável que as células apoptóticas não sejam eliminadas rapidamente no SNC e, portanto, sofram necrose secundária. [ 58 ] Por outro lado, a infecção por RV e, em particular, a superexpressão da proteína G de RV podem levar à piroptose, uma via de morte celular semelhante à apoptose que, ao contrário da apoptose, envolve a ativação da caspase 1 e, portanto, leva à necrose. [ 59 ] O grau de necrose ou piroptose induzido pela infecção por RV provavelmente desempenha um papel crítico na indução da imunidade antiviral. Enquanto as células apoptóticas mantêm a integridade da sua membrana e não estimulam a resposta imune inata, as células necróticas tornam-se permeabilizadas e secretam adjuvantes endógenos que podem desencadear uma resposta imune inata robusta. [ 60 ]
Como o nível de apoptose/necrose se correlaciona com a imunogenicidade do RV, sugere-se que o efeito imunoestimulatório das células apoptóticas/necróticas provavelmente contribui para a geração de uma resposta imune protetora. Portanto, a regulação da expressão do gene G do RV é muito provavelmente um fator importante na patogênese da raiva, pois fornece um meio para a sobrevivência e disseminação de variantes patogênicas do RV no sistema nervoso sem causar danos neuronais evidentes e desencadeando uma resposta imune protetora que impediria a infecção.
A expressão de RV G pode ser regulada no nível da síntese de RNA, no nível pós-traducional ou em ambos. Foi demonstrado que os níveis de RV G expressos por diferentes variantes quiméricas de RV são refletidos na taxa de síntese de RNA viral, sugerindo que a regulação diferencial da expressão de RV G por essas variantes resulta de variações na taxa de transcrição do mRNA viral. Assim como nas taxas de transcrição do RNA viral, a quantidade de RV G expressa por essas variantes se correlaciona inversamente com a patogenicidade viral. Por outro lado, a infecção de culturas neuronais primárias com a variante CVS-B2c menos patogênica de RV resultou em níveis quatro vezes maiores de proteína G do que a infecção com a variante CVS-N2c altamente patogênica, apesar da síntese de níveis comparáveis de mRNA de G em ambas as infecções. Experimentos de pulso-perseguição mostraram que os níveis mais altos de proteína G em neurônios infectados com CVS-B2c foram em grande parte o resultado de uma menor taxa de degradação da proteína G de CVS-B2c em comparação com a proteína G de CVS-N2c. Entretanto, o mecanismo que leva à degradação proteolítica mais rápida da proteína G CVS-N2c ainda precisa ser esclarecido.
Sintomas raiva
O período de incubação da raiva é, em média, de 30 a 90 dias. Em caso de infecção maciça por meio de grandes ferimentos na cabeça e no rosto, o período pode ser reduzido para 12 dias. Em casos raros, o período de incubação pode durar 1 ano ou mais.
Há uma mudança estritamente sequencial de três períodos da doença: prodrômico, excitação e paralisia.
O período prodrômico começa com o aparecimento de dor aguda ou em puxão no local da picada, bem como dor ao longo dos nervos. Na área da cicatriz, pode haver sensação de queimação, coceira, às vezes vermelhidão e inchaço. O paciente apresenta mal-estar geral, dor de cabeça e náuseas. Vômitos, aumento da temperatura corporal para 37,5-38 °C e sintomas de um transtorno mental progressivo são observados: aumento da excitabilidade reflexa, uma sensação inexplicável de ansiedade, medo, melancolia. Frequentemente, o paciente fica deprimido, inibido, retraído, recusa-se a comer, dorme mal, queixa-se de pensamentos sombrios e sonhos assustadores. O período prodrômico dura de 2 a 3 dias, às vezes se estende por até 7 dias. Ao final desse período, podem ocorrer crises de ansiedade com dificuldades respiratórias de curto prazo, sensação de aperto no peito, acompanhadas de taquicardia e aumento da frequência respiratória.
O período de excitação é marcado pelo aparecimento de hidrofobia: ao tentar beber, e então ao ver água ou ao se lembrar dela, o paciente experimenta um espasmo convulsivo da faringe e da laringe, durante o qual joga fora a caneca de água com um grito, joga as mãos trêmulas para a frente e joga a cabeça e o corpo para trás. O pescoço se estica, uma careta dolorosa distorce o rosto, que se torna azulado devido a um espasmo dos músculos respiratórios. Os olhos se arregalam, expressam medo, imploram por ajuda, as pupilas se dilatam e a inalação é difícil. No auge do ataque, pode haver parada cardíaca e respiratória. O ataque dura vários segundos, após os quais o estado do paciente parece melhorar. Posteriormente, ataques de espasmos dos músculos da laringe e da faringe podem ocorrer até mesmo com o movimento do ar (aerofobia), luz forte (fotofobia) ou uma palavra em voz alta (acusticofobia). Os ataques são acompanhados por agitação psicomotora, durante a qual o paciente se comporta como um "louco". A consciência fica turva durante a crise, mas melhora no período interictal. Durante o período de agitação, devido ao aumento do tônus do sistema nervoso simpático, os pacientes apresentam um aumento acentuado da salivação (sialorreia), com incapacidade de engolir saliva devido ao espasmo dos músculos faríngeos. O paciente borrifa saliva. Alguns pacientes podem desenvolver sinais de meningismo e até mesmo opistótono, e convulsões são comuns. Nesse caso, o líquido cefalorraquidiano pode não se alterar, mas em alguns pacientes, a concentração de proteínas pode aumentar e o número de células pode aumentar devido aos linfócitos.
Sem tratamento adequado, os sinais de desidratação aumentam, as feições faciais tornam-se mais acentuadas e o peso corporal diminui. A temperatura corporal atinge valores elevados. Convulsões são possíveis. A duração da fase de excitação é de cerca de 2 a 3 dias, raramente de 4 a 5 dias. Um desfecho fatal geralmente ocorre durante uma das crises. Raramente, o paciente sobrevive até o terceiro estágio da doença.
Durante o período de paralisia, o paciente se acalma. Os ataques de hidrofobia cessam, o paciente consegue beber e engolir alimentos, a consciência está lúcida. No entanto, apesar do aparente bem-estar, letargia, apatia e depressão aumentam, paralisia dos membros, distúrbios pélvicos e paralisia dos nervos cranianos logo aparecem. A temperatura corporal sobe para 42-43 °C, a pressão arterial cai e, ao final do primeiro dia, ocorre a morte por paralisia dos centros cardiovascular e respiratório.
Leucocitose neutrofílica, aumento de hemoglobina, eritrócitos e hematócrito são observados no sangue periférico.
O que está incomodando você?
Formulários
Clinicamente, distinguem-se as formas típicas e atípicas. As formas atípicas incluem todos os casos sem excitação e hidrofobia. As formas atípicas incluem bulbar, cerebelar, meningoencefalítica, etc.
Diagnósticos raiva
A detecção do antígeno rábico, anticorpos, RNA viral ou isolamento do vírus permite o diagnóstico da raiva. Como qualquer teste individual pode ser negativo em um paciente com raiva, amostras seriadas de soro para detecção de anticorpos antirrábicos, amostras de saliva para cultura viral e biópsia de pele para teste de imunofluorescência direta para antígeno viral são algumas vezes necessárias, especialmente quando há forte suspeita de raiva.
Um dos métodos mais rápidos para o diagnóstico antemortem da raiva em humanos é a realização de um teste de imunofluorescência direta em uma biópsia de pele da nuca para detectar o antígeno da raiva. O teste de imunofluorescência direta é o método mais sensível e específico para detectar o antígeno da raiva na pele e em outros tecidos frescos (por exemplo, biópsia cerebral), embora os resultados possam ocasionalmente ser negativos no início da doença. Se não houver tecido fresco disponível, a digestão enzimática de tecidos fixados pode aumentar a reatividade do teste de imunofluorescência; no entanto, a sensibilidade pode ser inaceitavelmente baixa.
O diagnóstico também pode ser estabelecido se o vírus for isolado da saliva após a inoculação de células de neuroblastoma ou de roedores de laboratório; isso geralmente é mais eficaz durante as primeiras 2 a 3 semanas da doença. A detecção de anticorpos neutralizantes do vírus da raiva, geralmente realizada pelo teste rápido de inibição de foco fluorescente (RFFIT), no soro de indivíduos não vacinados também é diagnóstica. A presença de anticorpos no líquido cefalorraquidiano confirma o diagnóstico, mas eles podem aparecer 2 a 3 dias depois dos anticorpos séricos e, portanto, podem ser menos úteis nos estágios iniciais da doença. Embora a resposta sorológica após a vacinação seja geralmente indistinguível da resposta sorológica induzida pela doença, a vacinação geralmente não produz anticorpos para o líquido cefalorraquidiano.
Apenas sete casos de "recuperação" da raiva nos últimos 25 anos foram bem documentados. Embora o vírus da raiva não tenha sido isolado de nenhum dos pacientes, altos títulos de anticorpos neutralizantes da raiva em amostras de soro e a presença de anticorpos neutralizantes no líquido cefalorraquidiano corroboraram fortemente o diagnóstico.
O que precisa examinar?
Quais testes são necessários?
Diagnóstico diferencial
O diagnóstico da raiva humana geralmente é feito com base em dados epidemiológicos e clínicos e confirmado em laboratório. O diagnóstico é simples se houver histórico de mordeduras de animais e se todo o espectro de sintomas e sinais tiver ocorrido. Caso contrário, uma avaliação cuidadosa, porém rápida, das características epidemiológicas e clínicas de casos menos típicos é necessária antes da realização de exames laboratoriais específicos. Qualquer paciente com sinais ou sintomas neurológicos ou encefalite inexplicada deve ser questionado sobre a possibilidade de exposição a animais em áreas endêmicas para raiva, dentro ou fora do país de residência. A falha em suspeitar de raiva em várias mortes humanas recentes nos Estados Unidos pode ter sido devido à falta de um histórico detalhado de exposição.
No início da doença, a raiva pode mimetizar muitas doenças infecciosas e não infecciosas. Muitas outras encefalites, como as causadas por herpesvírus e arbovírus, assemelham-se à raiva. Outras doenças infecciosas também podem mimetizar a raiva, como tétano, malária cerebral, riquetsiose e febre tifoide. Doenças infecciosas paralíticas que podem ser confundidas com a raiva incluem poliomielite, botulismo e encefalite por herpes símio B.
Doenças não infecciosas que podem ser confundidas com a raiva incluem diversas síndromes neurológicas, especialmente a polineuropatia inflamatória aguda (síndrome de Guillain-Barré), bem como encefalomielite alérgica pós-vacinação secundária à vacinação antirrábica do tecido nervoso, envenenamento ou intoxicação medicamentosa, abstinência alcoólica, porfiria aguda e histeria rábica. A síndrome de Guillain-Barré pode ser confundida com a raiva paralítica e vice-versa.
Quem contactar?
Tratamento raiva
O tratamento para a raiva não foi desenvolvido. A administração de altas doses de imunoglobulina antirrábica específica e interferon leucocitário é ineficaz. O tratamento sintomático é administrado para aliviar o sofrimento do paciente. Para isso, o paciente é colocado em uma enfermaria ou boxe separado, e é criado um regime de proteção que limita a influência do ambiente externo (ruído reduzido, luz forte, fluxo de ar). Para reduzir a excitabilidade do sistema nervoso central, são prescritos soníferos, anticonvulsivantes e analgésicos. O equilíbrio hídrico é normalizado.
Na fase paralítica, são prescritos medicamentos que estimulam a atividade dos sistemas cardiovascular e respiratório. Recomenda-se o uso de oxigenação hiperbárica, hipotermia cerebral e respiração mecânica controlada com curarização completa do paciente. No entanto, todos os métodos de tratamento são praticamente ineficazes. Na melhor das hipóteses, é possível prolongar a vida do paciente por vários meses. Um desfecho desfavorável é predeterminado pela gravidade do dano ao tronco encefálico, com a destruição de centros vitais.
Prevenção
O desenvolvimento da primeira vacina contra a raiva por Pasteur em 1885 inaugurou uma era de controle da raiva muito mais eficaz. Hoje, apesar da taxa de mortalidade de quase 100% em humanos por raiva, a doença é completamente prevenível por meio da vacinação pré e/ou pós-exposição. Enquanto Pasteur e seus colegas iniciaram a vacinação de cães particulares em Paris, a primeira vacinação em massa de cães foi realizada no início da década de 1920 no Japão, marcando o primeiro grande programa nacional de controle da raiva. A vacinação oral de animais selvagens, desenvolvida pela primeira vez na década de 1970, tem demonstrado repetidamente controlar a doença de forma eficaz em grandes hospedeiros terrestres, como raposas, guaxinins e gambás. [ 68 ] A vacinação antirrábica sustentada de populações de animais reservatórios com taxas de cobertura de 70% ou mais acabará por eliminar o RABV das espécies reservatório e impedir a disseminação do vírus para hospedeiros incidentais. [ 69 ]
Dados filogenéticos indicam que os lyssavírus infectaram morcegos muito antes de infectarem mamíferos terrestres, e a maioria dos lyssavírus, incluindo o RABV, ainda circula em várias espécies de morcegos em todo o mundo. [ 70 ] No entanto, métodos eficazes para prevenir a transmissão do RABV entre morcegos permanecem indefinidos, impossibilitando a possibilidade de erradicação completa da raiva neste momento. No entanto, mesmo após a exposição ao RABV através da mordida de um mamífero infectado pela raiva, a profilaxia pós-exposição segura e eficaz (PEP, incluindo limpeza de feridas, imunoglobulina antirrábica e vacinação antirrábica) pode proteger os humanos da infecção por raiva se o tratamento for administrado prontamente e de acordo com as recomendações da Organização Mundial da Saúde (OMS).
Esses dois métodos de prevenção de mortes humanas — um baseado na vacinação de pessoas expostas e o outro na vacinação de cães em número suficiente para interromper o ciclo de transmissão na fonte — constituem os alicerces de uma abordagem de "saúde única" para a prevenção e o controle da raiva canina. Esses dois meios distintos de prevenção de mortes humanas foram considerados como alternativas distintas: a Estratégia A, baseada no fornecimento de PEP às pessoas, e a Estratégia B, baseada na vacinação de cães; ou como componentes de uma Estratégia A + B combinada, em uma análise dos custos prováveis das estratégias alternativas.[ 71 ]
Países como a Tailândia tiveram enorme sucesso na prevenção de mortes humanas através da utilização da PEP, mas também encontraram uma procura crescente e custos associados à utilização apenas da PEP. [ 72 ] Por exemplo, em comparação com a situação em 1991, quatro vezes mais pessoas (mais de 400.000) necessitaram de PEP em 2003. Dados recentes mostram que a República Popular da China, que vacina 15 milhões de pessoas por ano após potencial exposição à raiva, gasta cerca de 650 milhões de dólares por ano apenas em PEP. [ 73 ]
Uma abordagem muito mais sustentável consiste em prevenir a propagação da infeção na fonte, na população animal, aumentando simultaneamente o acesso à PEP para doentes humanos expostos, quando necessário. Onde existe vontade política e financiamento adequado para controlar a raiva canina, as fatalidades podem ser e já foram eliminadas. A utilização generalizada da vacinação canina levou à eliminação da raiva canina em vários países, incluindo a Malásia em 1954, [ 74 ] o Japão em 1956, Taiwan em 1961, Singapura e, em particular, em toda a Europa Ocidental (revisto em Rupprecht et al, King et al, e Gongal e Wright). [ 75 ]
Использованная литература