Médico especialista do artigo

Novas publicações
Síndrome de Usher
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
A síndrome de Usher é uma doença hereditária que se manifesta como surdez completa desde o nascimento, bem como cegueira progressiva com a idade. A perda de visão está associada à retinite pigmentosa, um processo de degeneração pigmentar da retina. Muitas pessoas com síndrome de Usher também apresentam graves problemas de equilíbrio.
Epidemiologia
Graças à pesquisa, foi possível estabelecer que a síndrome de Usher afeta cerca de 8% das crianças surdo-mudas examinadas (os testes foram realizados em instituições especiais para surdo-mudos). Retinite pigmentar foi observada em 6 a 10% dos pacientes com surdez congênita, que, por sua vez, é observada em cerca de 30% das pessoas com doença pigmentar da retina.
Acredita-se que esta doença se manifeste em aproximadamente 3 a 10 pessoas em cada 100 mil em todo o mundo. Pode ser observada igualmente em mulheres e homens. Cerca de 5 a 6% da população mundial sofre desta síndrome. Cerca de 10% de todos os casos de surdez profunda na infância ocorrem devido à Síndrome de Usher tipo I, bem como aos tipos II.
Nos Estados Unidos, os tipos 1 e 2 são os mais comuns. Juntos, eles representam aproximadamente 90 a 95% de todos os casos de síndrome de Usher em crianças.
Causas Síndrome de Usher
A síndrome de Usher tipos I, II e III tem causa autossômica recessiva, enquanto a tipo IV é considerada uma doença do cromossomo X. As causas da cegueira e da surdez associadas a essa síndrome ainda não foram suficientemente estudadas. Acredita-se que as pessoas com essa doença sejam hipersensíveis a componentes que podem danificar a estrutura do DNA. Além disso, essa doença pode estar associada a distúrbios do sistema imunológico, mas, nesse caso, não há um quadro exato desse processo.
Em 1989, anormalidades cromossômicas foram identificadas pela primeira vez em pacientes com a doença do tipo II, o que pode, no futuro, levar a uma maneira de isolar os genes que causam a síndrome. Também pode ser possível identificar esses genes em portadores e desenvolver testes genéticos pré-natais específicos.
 [ 8 ]
[ 8 ]
Fatores de risco
A síndrome é herdada quando ambos os pais são afetados, ou seja, é herdada por um tipo recessivo. Uma criança também pode herdar a doença se seus pais forem portadores do gene. Se ambos os futuros pais tiverem esse gene, a probabilidade de ter um bebê com a síndrome é de 1 em 4. Uma pessoa que possui apenas um gene para a síndrome é considerada portadora, mas não apresenta sintomas do transtorno. Atualmente, ainda não é possível determinar se uma pessoa possui o gene para a doença.
Se uma criança nasce de pais que não têm esse gene, a probabilidade de ela herdar a síndrome é muito baixa, mas ela definitivamente será portadora.
Sintomas Síndrome de Usher
Os sintomas da síndrome de Usher incluem perda auditiva e acúmulo anormal de células pigmentadas nas estruturas oculares. O paciente desenvolve degeneração da retina, o que causa deterioração da visão e, nos casos mais graves, eventual perda da visão.
A perda auditiva neurossensorial pode ser leve ou completa e geralmente não progride desde o nascimento. No entanto, a doença pigmentar da retina pode começar a se desenvolver na infância ou mais tarde. Resultados de testes demonstraram que a acuidade visual central pode ser mantida por muitos anos, mesmo quando a visão periférica se deteriora (uma condição chamada "visão em túnel").
Estas são as principais manifestações da doença, que por vezes podem ser complementadas por outros distúrbios, como psicose e outros transtornos mentais, problemas no ouvido interno e/ou catarata.
Formulários
Durante a pesquisa, foram identificados 3 tipos desta doença, além de uma 4ª forma, que é bastante rara.
O tipo I da doença é caracterizado por surdez congênita completa, bem como por distúrbios do equilíbrio. Frequentemente, essas crianças começam a andar apenas com 1 ano e meio de idade. A deterioração da visão geralmente começa aos 10 anos de idade, e o desenvolvimento final do estado de cegueira noturna começa aos 20 anos. Crianças com esse tipo da doença podem desenvolver deterioração progressiva da visão periférica.
Na doença do tipo II, observa-se surdez moderada ou congênita. Nesse caso, a deterioração da surdez parcial geralmente não ocorre mais. A retinite pigmentar começa a se desenvolver por volta do final da adolescência ou após os 20 anos. O desenvolvimento da cegueira noturna geralmente começa entre 29 e 31 anos. O comprometimento da acuidade visual na patologia do tipo II geralmente progride um pouco mais lentamente do que no tipo I.
O tipo III da doença é caracterizado pela perda auditiva progressiva, geralmente com início na puberdade, bem como pelo desenvolvimento gradual durante o mesmo período (um pouco mais tarde que a perda auditiva) de retinite pigmentosa, que pode se tornar um fator no desenvolvimento de cegueira progressiva.
As manifestações da patologia do tipo IV ocorrem principalmente em homens. Nesse caso, também são observados distúrbios progressivos e perda de audição e visão. Essa forma é muito rara e geralmente tem natureza cromossômica X.
Diagnósticos Síndrome de Usher
O diagnóstico da síndrome de Usher é feito com base na combinação observada no paciente de surdez súbita e perda progressiva da visão.
Testes
Um teste genético especial pode ser solicitado para detectar a mutação.
Foram encontrados onze loci genéticos que podem causar o desenvolvimento da síndrome de Usher, e nove genes foram identificados como sendo definitivamente a causa do distúrbio:
- Tipo 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipo 2: ush2a, VLGR1, WHRN.
- Síndrome de Usher tipo 3: USH3A.
Cientistas do NIDCD, juntamente com colegas de universidades de Nova York e Israel, identificaram uma mutação chamada R245X no gene Pcdh15 que é responsável por uma grande porcentagem da síndrome de Usher tipo 1 na população judaica.
Para saber mais sobre laboratórios que realizam ensaios clínicos, visite https://www.genetests.org e pesquise no diretório de laboratórios por "síndrome de Usher".
Para saber mais sobre ensaios clínicos existentes que incluem testes genéticos para a síndrome de Usher, visite https://www.clinicaltrials.gov e pesquise por "síndrome de Usher" ou "teste genético para síndrome de Usher".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnóstico instrumental
Existem vários métodos de diagnóstico instrumental:
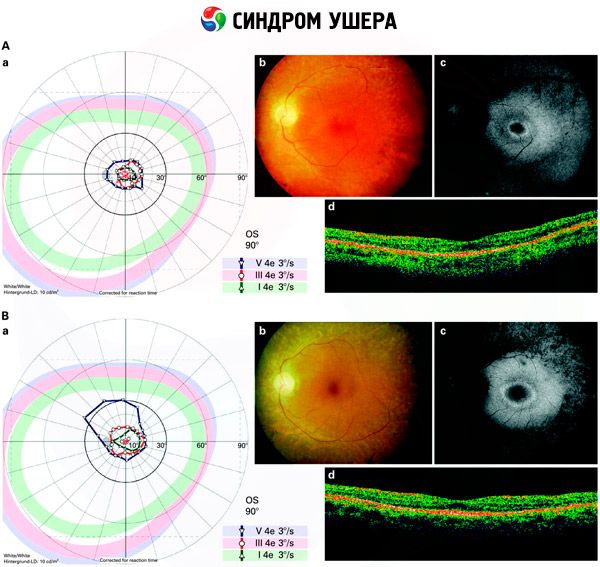
- Exame do fundo para detectar a presença de manchas pigmentares na retina, bem como estreitamento dos vasos retinianos;
- Eletrorretinograma, que permite detectar desvios degenerativos iniciais na retina. Mostra a extinção das vias eletrorradiográficas;
- Um eletronistagmograma (ENG) mede movimentos oculares involuntários que podem indicar a presença de um desequilíbrio.
- Audiometria, que é usada para determinar a presença de surdez e sua gravidade.
Diagnóstico diferencial
A síndrome de Usher deve ser diferenciada de alguns distúrbios semelhantes.
Síndrome de Hallgren, caracterizada por perda auditiva congênita e perda progressiva da visão (também podem ocorrer cataratas e nistagmo). Sintomas adicionais incluem ataxia, distúrbios psicomotores, psicose e retardo mental.
Síndrome de Alstrom, uma doença hereditária na qual a retina se degenera, resultando na perda da visão central. Esta síndrome está associada à obesidade infantil. Ao mesmo tempo, diabetes mellitus e perda auditiva começam a se desenvolver após 10 anos.
A rubéola em uma gestante no primeiro trimestre pode causar diversas anormalidades no desenvolvimento da criança. Entre as consequências de tal anormalidade estão a perda auditiva, bem como (ou) problemas de visão e, além disso, diversos defeitos de desenvolvimento.
Quem contactar?
Tratamento Síndrome de Usher
Atualmente, não há cura para a síndrome de Usher. Portanto, a terapia neste caso consiste principalmente em retardar o processo de perda da visão, bem como compensar a perda auditiva. Os possíveis métodos de tratamento incluem:
- Tomar vitamina A (alguns oftalmologistas acreditam que altas doses de palmitato de vitamina A podem retardar, mas não interromper, a progressão da retinite pigmentosa);
- Implantação de dispositivos eletrônicos especiais nos ouvidos do paciente (aparelhos auditivos, implantes cocleares).
Oftalmologistas recomendam que a maioria dos adultos com formas comuns de retinite pigmentosa tome 15.000 UI (unidades internacionais) de palmitato de vitamina A diariamente, sob supervisão. Como pessoas com síndrome de Usher tipo 1 não foram incluídas no estudo, altas doses de vitamina A não são recomendadas para esse grupo de pacientes. Pessoas que estejam considerando tomar vitamina A devem discutir essa opção de tratamento com seu médico. Outras recomendações para essa opção de tratamento incluem:
- Mudar sua dieta para incluir alimentos ricos em vitamina A.
- Mulheres que planejam engravidar devem parar de tomar altas doses de vitamina A três meses antes de planejarem engravidar devido ao aumento do risco de defeitos congênitos.
- Mulheres grávidas devem parar de tomar altas doses de vitamina A devido ao risco aumentado de defeitos congênitos.
Também é importante adaptar a criança à vida social. Isso requer o auxílio de professores de educação especial e psicólogos. Caso o paciente comece a apresentar perda progressiva da visão, ele deve ser ensinado a usar a linguagem de sinais.
Previsão
A síndrome de Usher tem um prognóstico desfavorável. O campo visual e sua acuidade começam a se deteriorar ao longo de 20 a 30 anos na maioria dos pacientes com esta doença, seja qual for o seu tipo. Em alguns casos, ocorre perda bilateral completa da visão. A perda auditiva, sempre acompanhada de mudez, evolui rapidamente para perda auditiva bilateral completa.