Médico especialista do artigo

Novas publicações
Síndrome de Aper
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.

 [
[ Causas Síndrome de Aper
O desenvolvimento da síndrome é provocado por um distúrbio na estrutura de um gene localizado no cromossomo 10. Esse gene é responsável pelo processo de separação dos dedos e, além disso, pelo fechamento oportuno das suturas cranianas.
Além disso, doenças infecciosas sofridas durante a gravidez (como rubéola ou tuberculose, sífilis ou gripe, bem como meningite) e a exposição da mãe a raios X são consideradas as causas da patologia. Na maioria das vezes, essa síndrome é detectada em crianças nascidas de pais idosos.
Patogênese
A síndrome de Apert é hereditária. Seu tipo é autossômico dominante (ou seja, em uma família em que um dos futuros pais é doente, a probabilidade de ter um bebê com a doença é de 50 a 100%).
Uma mutação única no receptor 2 do fator de crescimento de fibroblastos (FGFR2) resulta em um aumento no número de células progenitoras que se desenvolvem ao longo da via osteogênica. Isso, em última análise, resulta no aumento da formação da matriz óssea subperiosteal e na ossificação prematura das suturas cranianas durante o desenvolvimento fetal. A ordem e a taxa de fusão da sutura determinam o grau de deformidade e incapacidade. Uma vez que o material da sutura cicatriza, o crescimento de outros tecidos perpendiculares a essa sutura é limitado, e os ossos fundidos atuam como uma única estrutura óssea.
A primeira evidência genética de que a sindactilia na síndrome de Apert é causada por um defeito no receptor do fator de crescimento dos queratinócitos (KGFR) foi a observação de uma correlação entre a expressão do KGFR em fibroblastos e a gravidade da sindactilia.
Ambliopia e estrabismo são mais comuns em pacientes com a mutação FGFR2 Ser252Trp, e atrofia do disco óptico é mais comum em pacientes com a mutação FGFR2 Pro253Arg. Pacientes com mutações FGR2 Ser252Trp apresentam uma prevalência significativamente maior de deficiência visual em comparação com pacientes com a mutação FGFR2 Pro253Arg.
Sintomas Síndrome de Aper
Algumas manifestações da doença são claramente perceptíveis já no nascimento do bebê, pois se desenvolvem dentro do útero da mãe. Entre os principais sintomas da síndrome:
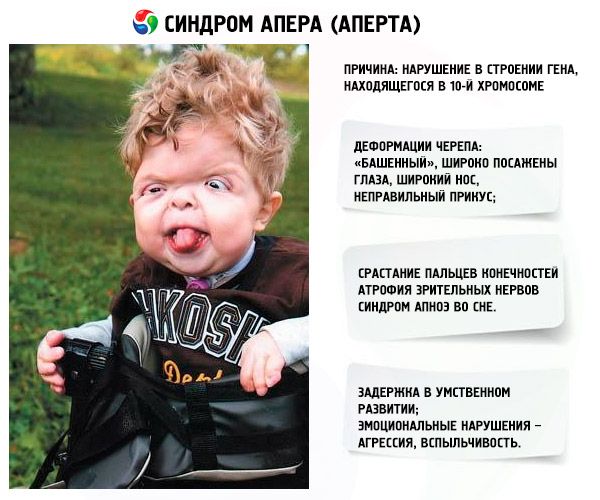
- O crânio é deformado - ele se estica em altura, adquirindo uma aparência de "torre"; além disso, os olhos são bem definidos e ligeiramente esbugalhados (porque o tamanho das órbitas oculares diminui); o nariz se alarga e uma mordida incorreta é formada (os dentes superiores se projetam excessivamente);
- Os dedos dos membros são completamente fundidos (principalmente o dedo anular, assim como o dedo médio e o indicador) e parecem uma membrana de pele ou uma fusão completa de ossos; além disso, dedos extras podem crescer;
- Retardo mental (não ocorre em todas as pessoas);
- Os nervos ópticos atrofiam, resultando em diminuição da acuidade visual (em alguns casos, ocorre perda completa da visão);
- O aumento da pressão intracraniana, que ocorre em decorrência do fechamento prematuro das suturas cranianas, manifesta-se na forma de dores de cabeça e vômitos com náuseas;
- Como o maxilar superior permanece subdesenvolvido, surgem problemas respiratórios;
- A síndrome da apneia do sono é uma ocorrência comum.
- Manifestações emocionais – agressividade, falta de controle, forte irascibilidade.
Complicações e consequências
A maioria dos pacientes apresenta algum grau de obstrução das vias aéreas superiores na infância. As vias aéreas superiores são pouco patenteadas devido ao tamanho reduzido da nasofaringe e das coanas, e as vias aéreas inferiores podem causar morte precoce devido a anormalidades da cartilagem traqueal.
Diagnósticos Síndrome de Aper
Para fazer um diagnóstico, são necessários os seguintes passos:
- O médico deve analisar as queixas do paciente, bem como o histórico médico. É necessário verificar se houve casos de patologia semelhante na família;
- Exame neurológico para avaliar a forma do crânio e o desenvolvimento intelectual do paciente (questionários especiais e também entrevista);
- Exame do fundo para detectar a presença de sintomas de aumento da pressão intracraniana (inchaço do disco óptico, bem como embaçamento de suas bordas);
- Para avaliar o estado do crânio, é realizado um raio X;
- Imagem computadorizada e de ressonância magnética da cabeça para examinar a estrutura do cérebro e do crânio camada por camada, para determinar a presença de sintomas de fusão prematura das suturas cranianas e, além disso, hidrocefalia (devido ao aumento da pressão intracraniana, o excesso de líquido cefalorraquidiano se acumula (este é o líquido cefalorraquidiano que promove o processo metabólico, bem como a nutrição do cérebro));
- Radiografia dos pés e das mãos para determinar a causa da fusão dos dedos (importante para o planejamento da intervenção cirúrgica subsequente);
- Pode ser prescrita uma consulta com um geneticista médico e um neurocirurgião.
Testes
A análise genética de mutações comuns que ocorrem no gene do tipo FGFR2 está sendo conduzida.
Diagnóstico diferencial
Esta síndrome deve ser diferenciada de outras patologias genéticas nas quais se observa craniossinostose. Trata-se de doenças como as síndromes de Pfeiffer, Crouzon, Saethre-Chotzen e Carpenter. Métodos de testes genéticos moleculares são utilizados para excluir essas anomalias.
Quem contactar?
Tratamento Síndrome de Aper
A cirurgia é considerada o único tratamento eficaz para a síndrome de Apert – ela ajuda a corrigir defeitos físicos individuais e também corrige o retardo mental.
Durante este procedimento, a sutura coronal é fechada para evitar possíveis lesões cerebrais. A técnica mais comum é a distração craniofacial, que envolve tração craniana gradual. A cirurgia ortodôntica e/ou ortognática é realizada para remover defeitos faciais individuais.
Além disso, os pacientes são submetidos à correção cirúrgica da fusão dos dedos.
Referências
Genética Médica. Guia Nacional. Editado por Ginter EK, Puzyrev VP GEOTAR-Media. 2022