Priões - agentes causadores das doenças causadas por priões
Last reviewed: 01.06.2018

Temos diretrizes rigorosas de fontes e só incluímos links para sites médicos fidedignos, instituições de investigação académica e, sempre que possível, estudos revistos por pares médicos. Note que os números entre parênteses ([1], [2], etc.) são ligações clicáveis para estes estudos.
Se achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável de alguma forma, selecione-o e prima Ctrl + Enter.
As infecções virais lentas são caracterizadas por critérios especiais:
- um período de incubação incomumente longo (meses, anos);
- uma lesão específica de órgãos e tecidos, principalmente do sistema nervoso central;
- progressão lenta e constante da doença;
- resultado fatal inevitável.

Alguns patógenos que causam infecções virais agudas também podem causar infecções virais lentas. Por exemplo, o vírus do sarampo às vezes causa SSPE, e o vírus da rubéola causa rubéola congênita progressiva e panencefalite rubeolar.
Uma infecção viral lenta típica em animais é causada pelo vírus Visna/Madi, que é um retrovírus. É o agente causador da infecção viral lenta e da pneumonia progressiva em ovinos. A substância branca do cérebro é destruída, desenvolve-se paralisia (visna - definhamento); ocorre inflamação crônica dos pulmões e do baço.
Doenças semelhantes em suas características às infecções virais lentas são causadas por príons – os agentes causadores das infecções por príons. As doenças por príons são um grupo de distúrbios progressivos do sistema nervoso central de humanos e animais. Em humanos, a função do sistema nervoso central é prejudicada, ocorrem alterações de personalidade e distúrbios do movimento. Os sintomas da doença geralmente duram de vários meses a vários anos, culminando em morte. Anteriormente, as infecções por príons eram consideradas juntamente com os chamados agentes causadores de infecções virais lentas.
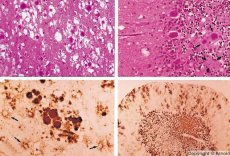
Alguns agentes causadores de doenças priônicas acumulam-se primeiro nos tecidos linfoides. Ao entrarem no cérebro, os príons acumulam-se em grandes quantidades, causando amiloidose (disproteinose extracelular, caracterizada pela deposição de amiloide com o desenvolvimento de atrofia e esclerose do tecido) e astrocitose (proliferação de neuroglia astrocítica, hiperprodução de fibras gliais). No cérebro, formam-se fibrilas, agregados proteicos ou amiloides e alterações espongiformes (encefalopatias espongiformes transmissíveis). Como resultado, ocorrem alterações comportamentais, comprometimento da coordenação motora e exaustão com desfecho fatal. A imunidade não é formada. As doenças priônicas são doenças conformacionais que se desenvolvem como resultado do dobramento incorreto (violação da conformação correta) de proteínas celulares necessárias para o funcionamento normal do corpo. As vias de transmissão dos príons são variadas:
- via alimentar - produtos de origem animal infectados, aditivos alimentares provenientes de órgãos bovinos crus, etc.:
- transmissão por transfusão de sangue, administração de medicamentos de origem animal, transplante de órgãos e tecidos, uso de instrumentos cirúrgicos e odontológicos infectados;
- transmissão através de preparações imunobiológicas (é conhecida a infecção de 1500 ovelhas com PrP''' pela vacina cerebral de formol de ovelhas doentes).
Os príons patológicos, ao entrarem no intestino, são transportados para o sangue e a linfa. Após a replicação periférica no baço, apêndice, amígdalas e outros tecidos linfoides, são transferidos para o cérebro através dos nervos periféricos (neuroinvasão). A penetração direta de príons no cérebro através da barreira hematoencefálica é possível. Anteriormente, acreditava-se que o sistema nervoso central era o único tecido no qual os príons patológicos se acumulavam, mas surgiram estudos que mudaram essa hipótese. Descobriu-se que o acúmulo de príons no baço está associado ao aumento e ao funcionamento das células dendríticas foliculares.
 [
[ Propriedades dos príons
A isoforma celular normal da proteína príon, com peso molecular de 33-35 kDa, é determinada pelo gene da proteína príon (o gene príon - PrNP está localizado no 20º cromossomo humano). O gene normal aparece na superfície celular (ancorado na membrana pela glicoproteína da molécula), sensível à protease. Regula a transmissão de impulsos nervosos, os ciclos diários, os processos de oxidação, participa do metabolismo do cobre no sistema nervoso central e da regulação da divisão das células-tronco da medula óssea. Além disso, o gene príon é encontrado no baço, nos linfonodos, na pele, no trato gastrointestinal e nas células dendríticas foliculares.
Proliferação de príons patológicos
A transformação de príons em formas alteradas ocorre quando o equilíbrio cineticamente controlado entre eles é rompido. O processo é intensificado pelo aumento da quantidade de príon patológico (PrP) ou exógeno. PrP é uma proteína normal ancorada na membrana celular. PrP' é uma proteína hidrofóbica globular que forma agregados consigo mesma e com PrP'' na superfície celular: como resultado, PrP' é transformado em PrP'' e então o ciclo continua. A forma patológica de PrP''' acumula-se nos neurônios, conferindo à célula uma aparência esponjosa.
Kuru
Doença priônica, anteriormente comum entre os papuas (que significa tremor ou tremor) na parte oriental da ilha da Nova Guiné. As propriedades infecciosas da doença foram comprovadas por K. Gajdusek. O patógeno é transmitido por alimentos como resultado de canibalismo ritual – comer o cérebro insuficientemente cozido e infectado por príons de parentes mortos. Como resultado de danos ao sistema nervoso central, os movimentos e a marcha são prejudicados, e surgem calafrios e euforia ("morte risonha"). O período de incubação dura de 5 a 30 anos. O paciente morre após um ano.
Doença de Creutzfeldt-Jakob
Doença priônica, que se manifesta como demência, distúrbios visuais e cerebelares e distúrbios do movimento, com desfecho fatal após 4 a 5 meses de doença na variante clássica da doença de Creutzfeldt-Jakob e após (3 a 14 meses na nova variante da doença de Creutzfeldt-Jakob). O período de incubação pode chegar a 20 anos. Diversas vias de infecção e causas da doença são possíveis:
- ao consumir produtos de origem animal insuficientemente tratados termicamente, como carne e cérebros de vacas com encefalopatia espongiforme bovina;
- durante o transplante de tecidos, como transplante de córnea, transfusão de sangue, uso de hormônios e outras substâncias biologicamente ativas de origem animal, uso de categute, instrumentos cirúrgicos contaminados ou insuficientemente esterilizados, manipulações prosetoriais;
- em caso de hiperprodução de PrR e outras condições que estimulem o processo de conversão de PrR' em PrR".
A doença também pode se desenvolver como resultado de uma mutação ou inserção na região do gene príon. A natureza familiar da doença é comum devido à predisposição genética para a doença de Creutzfeldt-Jakob. Na nova variante da doença de Creutzfeldt-Jakob, os distúrbios se desenvolvem em uma idade mais jovem (idade média de 28 anos), em contraste com a variante clássica (idade média de 65 anos). Na nova variante da doença de Creutzfeldt-Jakob, a proteína príon anormal se acumula não apenas no sistema nervoso central, mas também nos tecidos linforreticulares, incluindo as amígdalas.
Síndrome de Gerstmann-Sträussler-Scheinker
Doença hereditária por príons, acompanhada de demência, hipotonia, distúrbio de deglutição (disfagia) e disartria. Frequentemente tem natureza familiar. O período de incubação é de 5 a 30 anos. A doença ocorre entre 50 e 60 anos e sua duração varia de 5 a 13 anos.
Insônia fatal hereditária
Doença autoimune com insônia progressiva, hiper-reatividade simpática (hipertensão, hipertermia, hiperidrose, taquicardia), tremor, ataxia, multideficiência e alucinações. O sono é gravemente perturbado. A morte ocorre com a progressão da insuficiência cardiovascular.
Raspar
Scrapie (do inglês scrape - raspar) é uma doença priônica de ovinos e caprinos (sarna), que ocorre com danos ao sistema nervoso central, distúrbios progressivos do movimento, coceira intensa na pele (sarna) e termina na morte do animal.
Encefalopatia espongiforme bovina
Doença bovina caracterizada por danos ao sistema nervoso central, coordenação prejudicada dos movimentos e morte inevitável do animal. A epidemia da doença eclodiu inicialmente na Grã-Bretanha. Foi associada à alimentação de animais com farinha de carne e ossos contendo príons patológicos. O período de incubação varia de 1,5 a 15 anos. O cérebro, a medula espinhal e os globos oculares dos animais são os mais infectados.
Diagnóstico laboratorial de doenças priônicas
Durante o diagnóstico, observam-se alterações espongiformes no cérebro, astrocitose (gliose) e ausência de infiltrados inflamatórios. O cérebro é corado para amiloide. Marcadores proteicos de distúrbios cerebrais priônicos são detectados no líquido cefalorraquidiano (por ELISA). É realizada a análise genética do gene priônico (PCR).
Prevenção de doenças priônicas
Autoclavagem (a 134 °C por 18 min; a 121 °C por 1 h), incineração, tratamento adicional com água sanitária e solução de NaCl mono-normal por 1 h são recomendados para a descontaminação de instrumentos e objetos do ambiente. Para profilaxia não específica, foram introduzidas restrições ao uso de medicamentos de origem animal e a produção de hormônios hipofisários de origem animal é proibida. O transplante da dura-máter é restrito. Luvas de borracha são utilizadas ao trabalhar com fluidos dialógicos de pacientes.