Médico especialista do artigo

Novas publicações
Mucopolissacaridose tipo 3
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
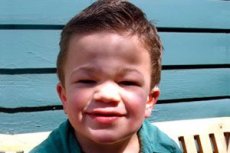
Mucopolissacaridose tipo III (sinônimos: síndrome de Sanfilippo, deficiência de aN-acetilglucosaminidase lisossomal - mucopolissacaridose III A, deficiência de acetil-CoA-a-glucosaminida-N-acetiltransferase - mucopolissacaridose III B, N-acetilglucosamina-6-sulfatase - mucopolissacaridose III C, deficiência de sulfamidase - mucopolissacaridose III D).
 [
[ Patogênese
A doença é causada por mutações em quatro genes diferentes: aN-acetilglicosaminidase lisossomal (mucopolissacaridose III A), acetil-CoA-a-glicosaminida-N-acetiltransferase (mucopolissacaridose III B), N-acetilglicosamina-6-sulfatase lisossomal (mucopolissacaridose III C) e sulfamidase (mucopolissacaridose III D). Todas as enzimas estão envolvidas no metabolismo do sulfato de heparana.
O gene da heparan-N-sulfatase - SGSH - está localizado no braço longo do cromossomo 17 - 17q25.3. 75,3% das mutações atualmente conhecidas no gene SGSH são mutações pontuais. Mutações frequentes características de populações europeias foram descritas - R74C (56% na Polônia e 21% na Alemanha) e R245H (56% na Holanda).
A frequência da mutação R74C é de 47,5%, enquanto a da mutação R245H é de 7,5%. As outras duas mutações descritas, delll35G e N389S, juntas representam 21,7% dos alelos mutantes.
O gene da aN-acetil-glicosaminidase (NAGLU) está localizado no braço longo do cromossomo 17 - 17q21. 69% das mutações encontradas no gene NAGLU são mutações sem sentido e sem sentido, e 26,3% são pequenas deleções e inserções. O gene da acetil-CoA-cc-glicosaminidase-N-acetiltransferase (HGSNAT) está localizado no braço curto do cromossomo 8 - 8p11.1. O gene foi caracterizado apenas em 2006 e, até o momento, apenas algumas mutações foram encontradas nele.
O gene da N-acetil-glucosamina-6-sulfatase - GNS - está localizado no braço longo do cromossomo 12 - 12ql4. Há 12 pacientes com mucopolissacaridose IIID registrados no mundo. Quatro mutações no gene GNS foram descritas.
Em todos os subtipos de mucopolissacaridose III, há um distúrbio na degradação do sulfato de heparana, que faz parte da estrutura das membranas celulares, incluindo membranas neuronais, o que se correlaciona com um processo neurodegenerativo grave causado por atrofia cortical. A diarreia crônica é explicada pelo envolvimento do sistema nervoso autônomo no processo patológico, juntamente com a disfunção da mucosa intestinal. A perda auditiva neurossensorial é provavelmente devida a três causas: otite frequente, deformação dos ossículos auditivos e anomalias do ouvido interno. A rigidez articular é o resultado da deformação das metáfises, o espessamento da cápsula articular é secundário à deposição de glicosaminoglicanos e fibrose. As diferenças intrassindrômicas na gravidade da doença são devidas exclusivamente à atividade funcional residual da enzima mutante: quanto maior, mais branda a doença.
Sintomas mucopolissacaridose tipo 3
O polimorfismo clínico na síndrome de Sanfilippo é menos pronunciado do que em outros tipos de mucopolissacaridose. A progressão lenta da doença e distúrbios neurológicos graves com sintomas leves em órgãos internos e outros sistemas são características.
Os primeiros sintomas da doença geralmente aparecem entre 2 e 6 anos de idade em crianças com desenvolvimento normal. Os sintomas manifestos incluem regressão do desenvolvimento psicomotor e da fala, transtornos psiquiátricos na forma de síndrome de hiperatividade, comportamento autista ou agressivo, distúrbios do sono; as crianças tornam-se descuidadas e desatentas.
Outros sintomas comuns são hirsutismo, pelos grossos, hepatoesplenomegalia moderada, deformidade em valgo dos membros e pescoço curto. O desenvolvimento de características faciais grosseiras, como gargoilismo, e deformidades esqueléticas, como disostose múltipla, são fracamente expressos na mucopolissacaridose III, em comparação com outros tipos de mucopolissacaridose caracterizados pelo fenótipo de Hurler. A altura, em geral, corresponde à idade, e a rigidez articular raramente causa disfunção. A maioria dos pacientes frequentemente desenvolve osteoporose e osteomalacia. Distúrbios esqueléticos secundários - alto risco de fraturas patológicas. Distúrbios psiconeurológicos graves são mais frequentemente observados entre o 6º e o 10º ano de vida e levam a um desajustamento social pronunciado. A perda auditiva neurossensorial progressiva é inerente a todos os pacientes com formas graves e moderadas da doença. Convulsões são observadas em quase todos os pacientes à medida que a doença progride.
A doença progride rapidamente, e a maioria dos pacientes não sobrevive até os 20 anos. A mucopolissacaridose IIIA é considerada o tipo mais comum e grave dessa síndrome.
Diagnósticos mucopolissacaridose tipo 3
O diagnóstico de mucopolissacaridose III é confirmado pela determinação do nível de excreção urinária de glicosaminoglicanos e pela medição da atividade enzimática. No caso da mucopolissacaridose III, a excreção total de glicosaminoglicanos na urina aumenta e observa-se hiperexcreção de sulfato de heparana. A atividade das enzimas lisossomais correspondentes a um subtipo específico de mucopolissacaridose III é medida em leucócitos ou em cultura de fibroblastos cutâneos utilizando um substrato fluorogênico artificial.
O diagnóstico pré-natal é possível medindo a atividade enzimática na biópsia de vilo corial entre 9 e 11 semanas de gestação e/ou determinando o espectro de glicosaminoglicanos no líquido amniótico entre 20 e 22 semanas de gestação. Para famílias com genótipo conhecido, o diagnóstico de DNA pode ser realizado no início da gestação.
Quais testes são necessários?
Diagnóstico diferencial
O diagnóstico diferencial é realizado tanto dentro do grupo das mucopolissacaridoses quanto com outras doenças de depósito lisossomal: mucolipidoses, galactosialidose, sialidose, manosidose, fucosidose, gangliosidose GM1.
Quem contactar?
Tratamento mucopolissacaridose tipo 3
Até o momento, não foram desenvolvidos métodos eficazes de tratamento para mucopolissacaridose III. O tratamento sintomático é indicado.
Использованная литература