Médico especialista do artigo

Novas publicações
Síndrome de Pierre Robin
Última revisão: 04.07.2025

Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.

A síndrome de Pierre Robin, também conhecida na medicina como anomalia de Robin, é uma patologia congênita do desenvolvimento da parte maxilar da face. A doença recebeu esse nome em homenagem ao dentista francês P. Robin, que primeiro descreveu todos os seus sinais. Lannelongue e Menard descreveram a síndrome de Pierre Robin pela primeira vez em 1891, em seu relato sobre dois pacientes com micrognatia, fenda palatina e retroglossoptose. Em 1926, Pierre-Robin publicou um caso da doença em uma criança com sinais da síndrome clássica. Até 1974, a tríade de sinais era conhecida como síndrome de Robin-Pierre. No entanto, essa síndrome agora é usada para descrever malformações com a presença simultânea de múltiplas anomalias.
Epidemiologia
É um defeito congênito heterogêneo com prevalência de 1 em 8.500 nascidos vivos. A proporção entre homens e mulheres é de 1:1, exceto na forma ligada ao cromossomo X.
Entre esses pacientes, 50% dos bebês têm fenda incompleta do palato mole, o restante nasce com palato arqueado e anormalmente alto, mas sem fenda.
Causas Síndrome de Pierre Robin
A possibilidade de herança autossômica recessiva da doença é considerada. Existem dois tipos de síndrome, dependendo da etiologia: isolada e geneticamente determinada. O tipo isolado se desenvolve devido à compressão da parte inferior da mandíbula durante o desenvolvimento embrionário. A compressão pode se desenvolver devido a:
- Presença de selos localizados no útero (cistos, cicatrizes, tumores).
- Gravidez múltipla.
Além disso, o desenvolvimento da mandíbula no feto pode ser interrompido por:
- Infecções virais que a futura mãe sofreu durante a gravidez.
- Distúrbios neurotróficos.
- Quantidade insuficiente de ácido fólico no corpo de uma mulher grávida.
Patogênese
A síndrome de Pierre Robin é causada por distúrbios embrionários causados por uma grande variedade de patologias no período pré-natal.
Existem três teorias fisiopatológicas que podem explicar a ocorrência da síndrome de Pierre Robin.
Teoria Mecânica: Esta teoria é a mais provável. O subdesenvolvimento do aparelho mandibular ocorre entre a 7ª e a 11ª semanas de gestação. A posição alta da língua na cavidade oral leva à formação de fendas no palato, impedindo o fechamento da veia cava. Esta teoria explica a clássica fenda em U invertido e a ausência do lábio leporino associado. Oligoidrâmnio pode desempenhar um papel na etiologia, visto que a ausência de líquido amniótico pode levar à deformação do queixo e à subsequente compressão da língua entre as veias cavas.
Teoria neurológica: Atraso no desenvolvimento neurológico foi observado na eletromiografia dos músculos da úvula e da coluna faríngea, e no paladar devido ao atraso na condução do nervo hipoglosso.
Teoria da disneurorregulação do rombencéfalo: Esta teoria é baseada na interrupção do desenvolvimento do rombencéfalo durante a ontogênese.
O desenvolvimento insuficiente da parte inferior da mandíbula da criança leva a uma redução significativa da cavidade oral. Isso, por sua vez, causa a chamada pseudomacroglossia, ou seja, a língua é deslocada para trás da parede faríngea. Essa patologia leva ao desenvolvimento de obstrução das vias aéreas.
Enquanto o bebê chora ou se move, as vias aéreas permanecem desobstruídas, mas assim que o bebê adormece, a obstrução ocorre novamente.
Devido a distúrbios respiratórios, o processo de alimentação do bebê é muito difícil. Nesse período, quase sempre ocorre obstrução das vias aéreas. Se não houver correção médica, essa patologia pode levar à exaustão grave de todo o corpo e até à morte.
Sintomas Síndrome de Pierre Robin
A doença é caracterizada por três sintomas principais:
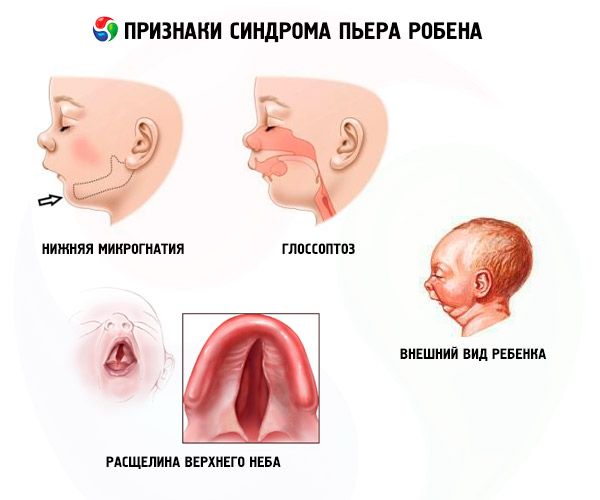
- Micrognatia inferior (subdesenvolvimento do maxilar inferior, ocorre em 91,7% dos casos da doença). Caracteriza-se pela retração da arcada dentária inferior em 10 a 12 mm em relação à arcada superior. A arcada inferior tem um corpo pequeno, com um ângulo obtuso. A criança atinge o desenvolvimento normal por volta dos 5 a 6 anos de idade.
- Glossoptose (retração da língua devido ao seu desenvolvimento insuficiente, observada em 70-85% dos casos).
- Macroglossia e anquiloglossia são sintomas relativamente raros, observados em 10-15% dos casos.
- Uma rachadura aparece no céu.
- Bradipneia e dispneia.
- Cianose leve.
- Asfixia, que ocorre mais frequentemente durante tentativas de alimentar o bebê.
- Engolir é impossível ou muito difícil.
- Sentindo vontade de vomitar.
- Anomalias auriculares em 75% dos casos.
- Perda auditiva condutiva ocorre em 60% dos pacientes, enquanto atresia do conduto auditivo externo ocorre em apenas 5% dos pacientes, pneumatização insuficiente da cavidade mastoide do osso temporal.
- Anomalias do ouvido interno (aplasia dos canais semicirculares laterais, grande aqueduto vestibular, perda de células ciliadas da cóclea).
- Malformações nasais são incomuns e consistem principalmente em anomalias da raiz nasal.
- Malformações dentárias ocorrem em 30% dos casos. Laringomalácia e insuficiência velofaríngea ocorrem em aproximadamente 10 a 15% dos pacientes com síndrome de Pierre Robin.
Características sistêmicas da síndrome de Pierre Robin
Anomalias sistêmicas do desenvolvimento são descritas em 10-85% dos casos registrados.
Anormalidades oculares ocorrem em 10 a 30% dos pacientes. Elas podem incluir: hipermetropia, miopia, astigmatismo, esclerose corneana e estenose do ducto nasolacrimal.
Patologias cardiovasculares: sopros cardíacos benignos, estenose da artéria pulmonar, persistência do canal arterial, janela oval, comunicação interatrial e hipertensão pulmonar. Sua prevalência varia de 5% a 58%.
Anomalias relacionadas ao sistema musculoesquelético (70-80% dos casos): sindactilia, falanges displásicas, polidactilia, clinodactilia, hipermobilidade articular e oligodactilia dos membros superiores. Anomalias dos membros inferiores: anomalias dos pés (pé torto, adução metatarsiana), malformações femorais (pelve em valgo ou varo, fêmures curtos), anomalias do quadril (luxação congênita, contraturas), anomalias da articulação do joelho (GENO VALGO, sincondrose). Malformações da coluna vertebral: escoliose, cifose, lordose, displasia vertebral, agenesia do sacro e do seio coccígeo.
Patologias do sistema nervoso central: epilepsia, atrasos no desenvolvimento do sistema nervoso, hidrocefalia. A frequência de defeitos do SNC é de cerca de 50%.
Anomalias geniturinárias: testículos não descidos (25%), hidronefrose (15%) e hidrocele (10%).
Síndromes e condições associadas: síndrome de Stickler, síndrome da trissomia 11q, trissomia 18, síndrome de deleção 4q, artropatia reumatoide, hipocondroplasia, síndrome de Moebius.
Estágios
Existem três estágios de gravidade da doença, que dependem da condição do trato respiratório da criança:
- Leve - há pequenos problemas com a alimentação, mas a respiração quase não é difícil. O tratamento é realizado em regime ambulatorial.
- Moderado – a respiração é moderadamente difícil, a alimentação da criança é moderadamente difícil. O tratamento é realizado em um hospital.
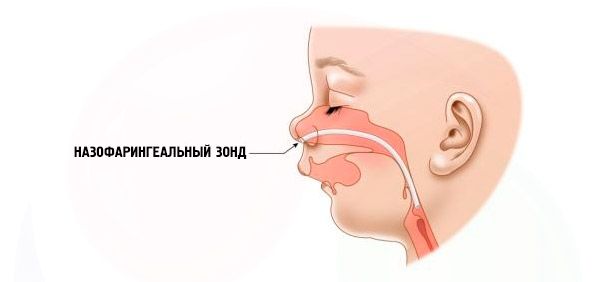
- Grave – a respiração fica muito difícil, a criança não consegue se alimentar normalmente. É necessário o uso de dispositivos especiais (sonda intranasal).
Complicações e consequências
A combinação de micrognatia e glossoptose pode levar a complicações respiratórias graves e problemas durante a alimentação da criança.
A síndrome de Pierre Robin causa as seguintes complicações:
- Respiração estridosa devido à obstrução das vias aéreas. Laringomalácia ou até mesmo asfixia do sono.
- O desenvolvimento psicomotor da criança fica muito atrás do de seus pares.
- O desenvolvimento físico também fica para trás.
- A fala dos pacientes fica prejudicada.
- Infecções de ouvido frequentes que se tornam crônicas e levam à deficiência auditiva.
- Síndrome da apneia obstrutiva do sono, a ocorrência de morte durante o sono varia em 14-91% dos casos.
- Problemas com os dentes.
Diagnósticos Síndrome de Pierre Robin
O diagnóstico da síndrome de Pierre Robin não é difícil. Baseia-se nas manifestações clínicas. Para descartar outras patologias, é muito importante consultar um geneticista.
Crianças com anomalia congênita de Robin apresentam problemas respiratórios desde o nascimento devido à língua constantemente afundada para trás. O bebê fica inquieto, sua pele fica azulada e há chiado no peito ao inspirar. Pode ocorrer engasgo durante a alimentação. O diagnóstico também pode ser feito pela aparência incomum da criança – "cara de pássaro". Frequentemente, os pacientes desenvolvem outras anomalias: miopia, catarata, patologia do aparelho geniturinário, patologia cardíaca e anomalias no desenvolvimento da coluna vertebral.
Com base nessas manifestações clínicas, não será difícil para um especialista fazer um diagnóstico correto.
Quem contactar?
Tratamento Síndrome de Pierre Robin
O tratamento é realizado imediatamente após o nascimento de uma criança com síndrome de Pierre Robin. Se a doença for leve, para melhorar o estado do paciente, é necessário manter a criança constantemente na vertical ou deitada de bruços. A cabeça do bebê deve estar inclinada em direção ao peito. Durante a alimentação, não é recomendado manter a criança na horizontal para que o alimento não entre nas vias aéreas.
Se o subdesenvolvimento do maxilar inferior for bastante pronunciado, a intervenção cirúrgica é utilizada para trazer a língua retraída para uma posição fisiológica normal. Em casos graves, a língua é puxada para cima e fixada no lábio inferior. Em casos muito graves, traqueostomia, glossopexia e distração osteogênica do maxilar inferior devem ser realizadas.
O tratamento conservador também é utilizado.
Medicação
Fenobarbital. Um comprimido para dormir e sedativo, com efeito anticonvulsivante. Cada comprimido contém 100 ml de fenobarbital. A dosagem é individual, pois depende da gravidade da doença e do estado da criança. O medicamento é proibido para pacientes com insuficiência hepática, hipercinesia, anemia, miastenia gravis, porfiria, diabetes mellitus, depressão e intolerância aos componentes. Os seguintes sintomas são possíveis ao tomá-lo: tontura, astenia, alucinações, agranulocitose, náusea, pressão arterial baixa e alergias.
Clonazepam. Medicamento prescrito para o tratamento da epilepsia. Contém como substância ativa o clonazepam, um derivado benzodiazepínico. Possui efeitos anticonvulsivantes, ansiolíticos e relaxantes musculares. A dose é determinada pelo médico assistente, mas não deve exceder o máximo de 250 mcg por dia. Não tome em caso de insônia, hipertonia muscular, agitação psicomotora ou transtornos de pânico. Os seguintes sintomas são possíveis durante o uso: letargia, náusea, dismenorreia, dor de cabeça, leucopenia, retenção ou incontinência urinária, alopecia e alergias.
Sibazon. Disponível na forma de solução e comprimidos retais. O ingrediente ativo é um derivado benzodiazepínico (sibazon). Possui efeito sedativo, ansiolítico e anticonvulsivante. A dosagem é individual. Pacientes com hipercapnia crônica, miastenia gravis ou intolerância a benzodiazepínicos não podem tomar o medicamento. Ao usar o medicamento, podem ocorrer os seguintes sintomas: náusea, constipação, dor de cabeça, tontura, soluços, incontinência urinária e alergias.
Liofilizado de cortexina. Medicamento com efeito nootrópico. Contém um complexo de frações polipeptídicas hidrossolúveis e glicina. A dosagem é individual e prescrita pelo médico assistente, de acordo com a condição do paciente. Pacientes com intolerância à cortexina não podem tomar este medicamento. O medicamento pode causar reações alérgicas.
Tratamento de fisioterapia
Normalmente, em estágios leves da síndrome, é usada a terapia posicional, na qual a criança é colocada de bruços, na posição vertical, até que a gravidade force o maxilar inferior a crescer corretamente.
Tratamento cirúrgico
O tratamento cirúrgico é usado principalmente para corrigir a glossoptose. Existem vários métodos:
- Suporte da língua com fio de prata. O fio é passado através da parte inferior da gengiva e do lábio inferior. O método é chamado de Douglas.
- Método de Duhamel: um fio grosso de prata é passado pela base da língua e ambas as bochechas do paciente. Usar por no máximo trinta dias.
- Aparelhos ortopédicos para extensão e fixação da língua.
- Com um ano de idade, a cirurgia para corrigir a fenda palatina pode ser realizada.
Previsão
O prognóstico e a evolução da doença são graves. Na maioria das vezes, a morte ocorre nos primeiros dias de vida, nos estágios moderado e grave da doença (a causa é asfixia). Além disso, o risco de morte no primeiro ano é bastante alto devido a inúmeras infecções.
Para pacientes com mais de dois anos de idade, o prognóstico é favorável.
 [ 36 ]
[ 36 ]